The chemical dataset curation function is implemented in Chem features.
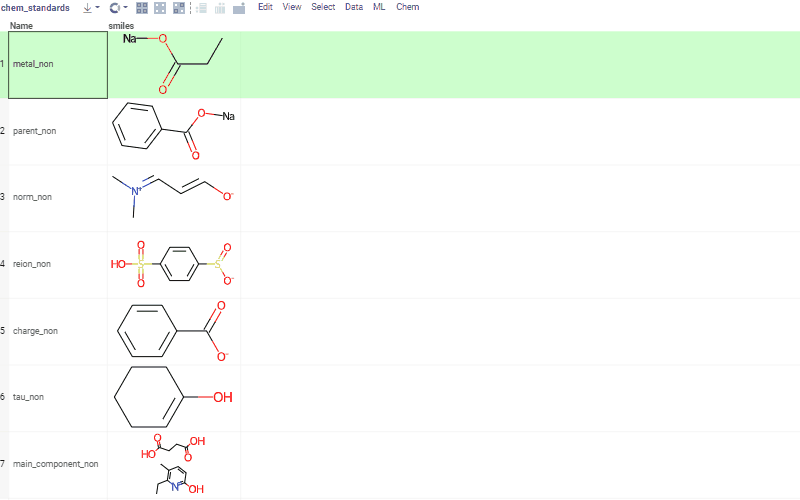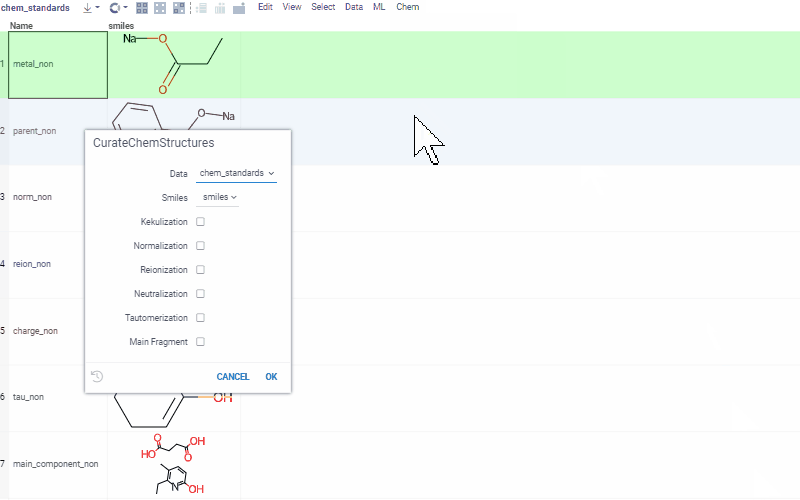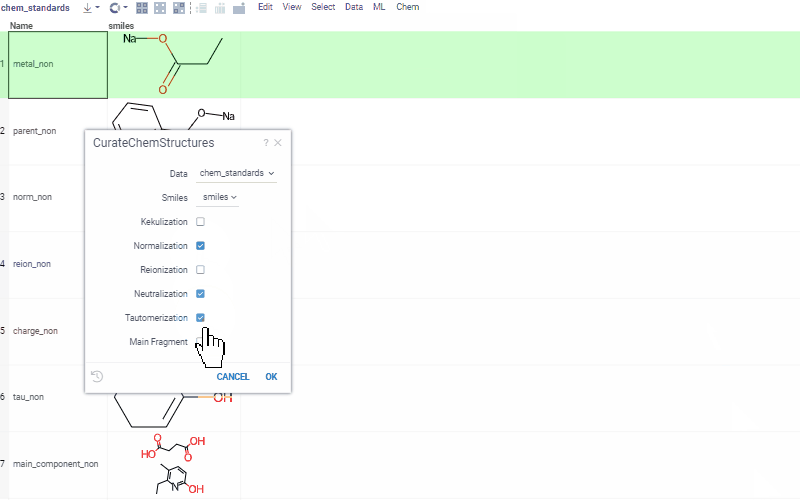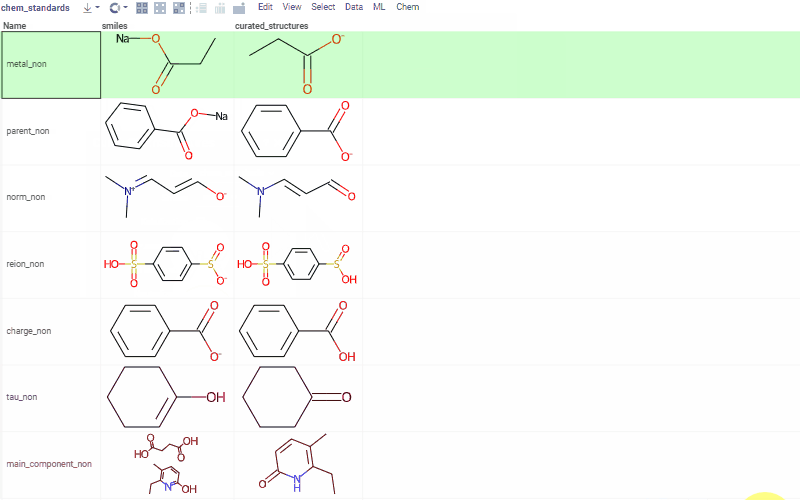
Curation tools include, but are not limited to:
- kekulization
- normalization
- neutralization
- tautomerization
- selection of the main component
See Chemical dataset curation for more details, and a demo with curation examples.