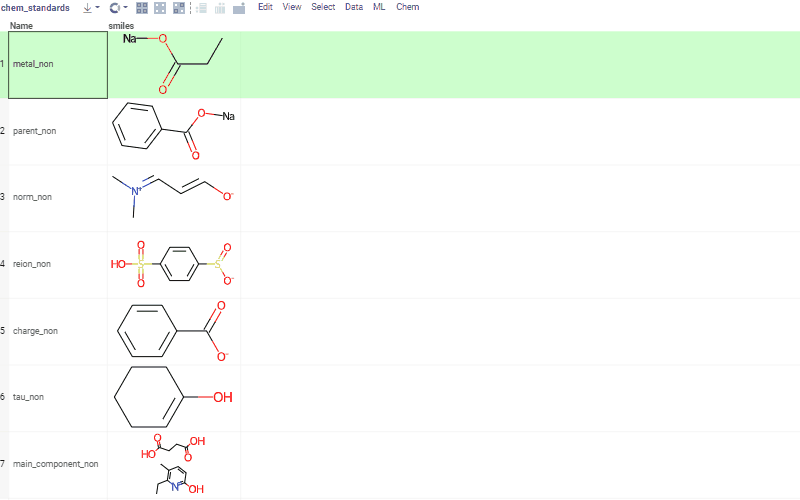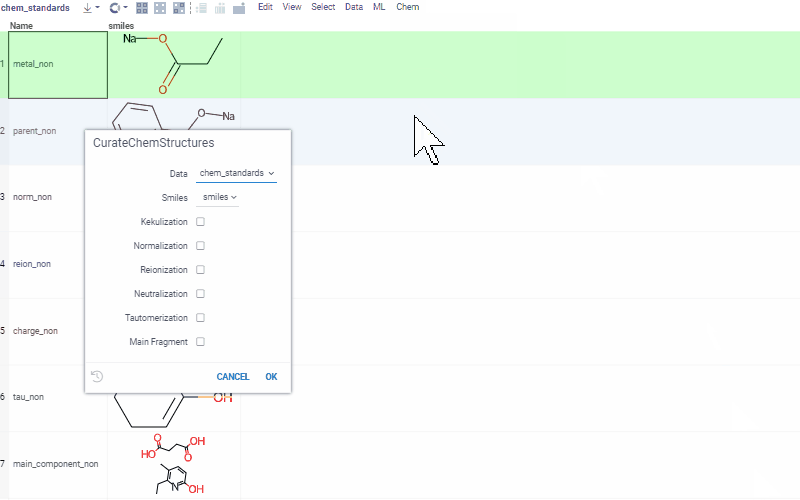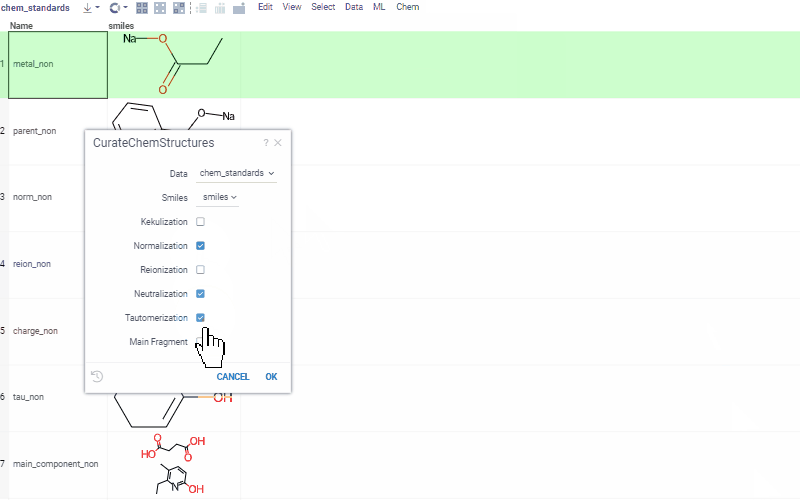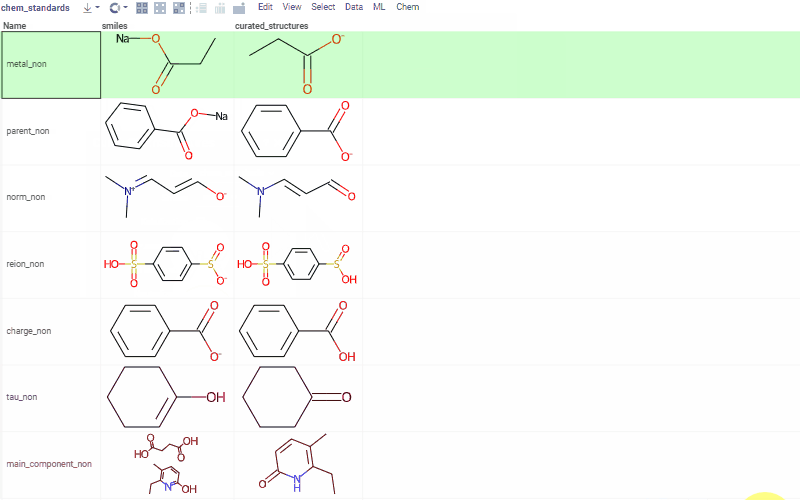
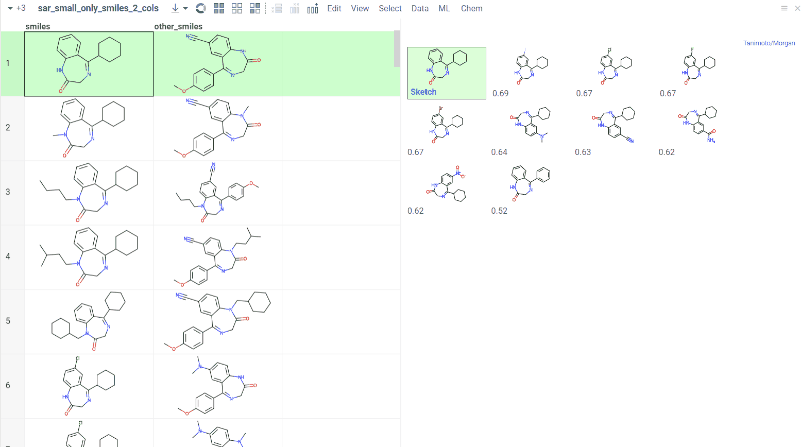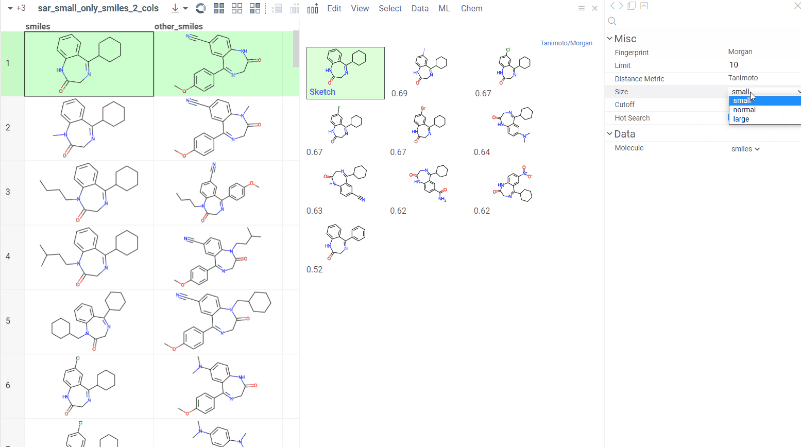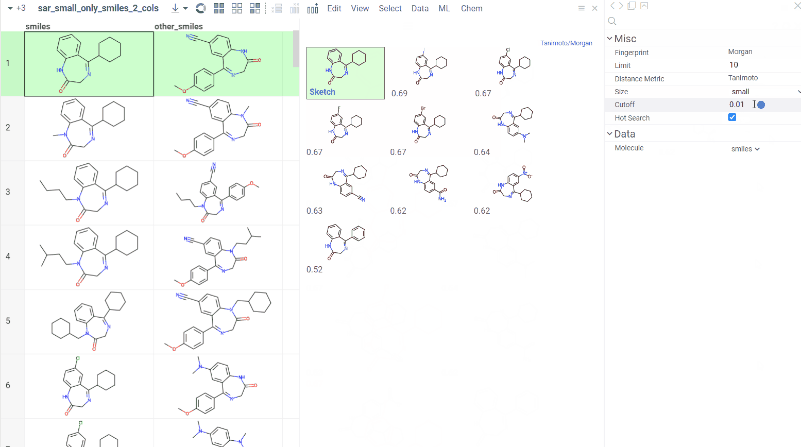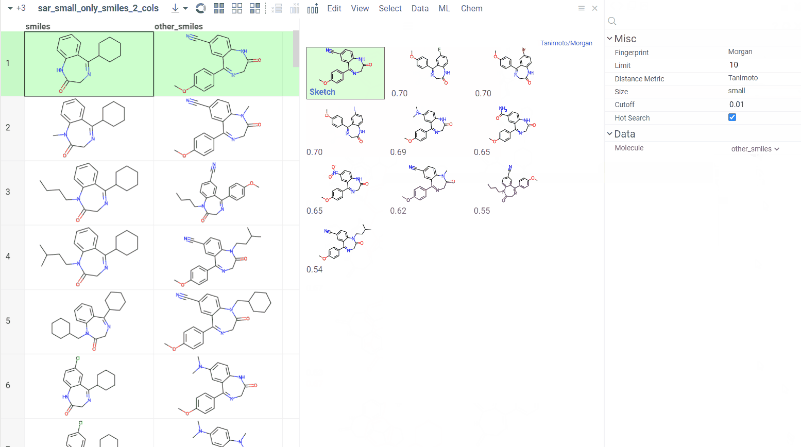
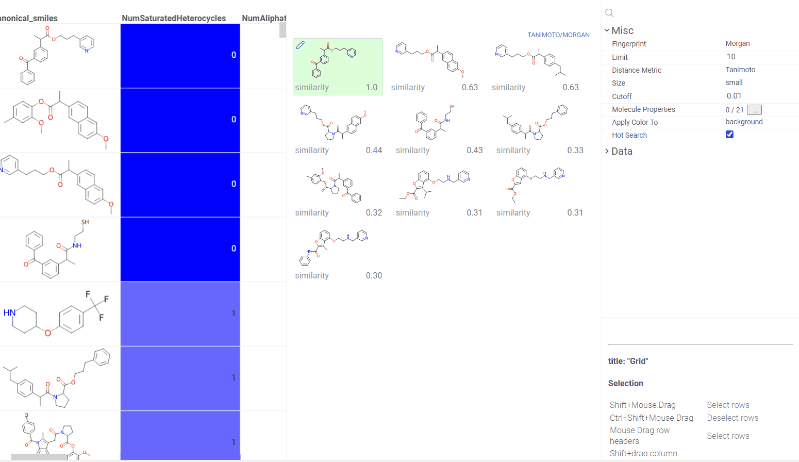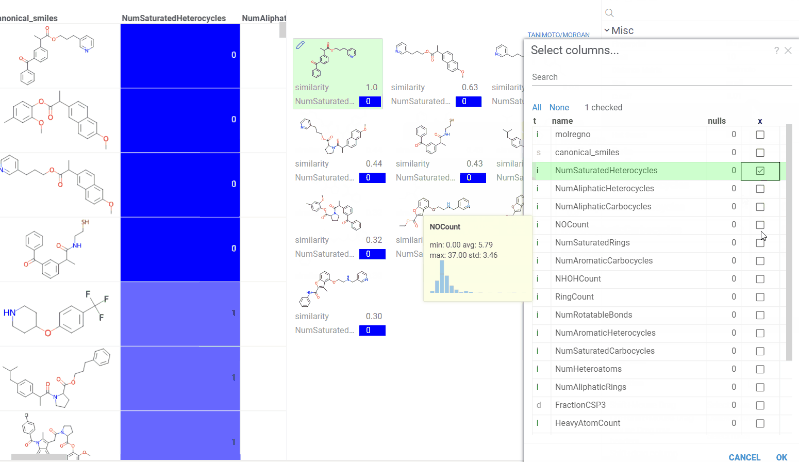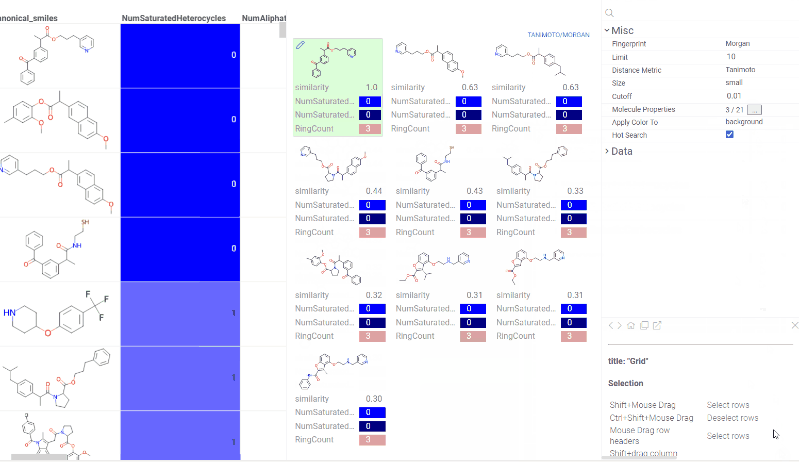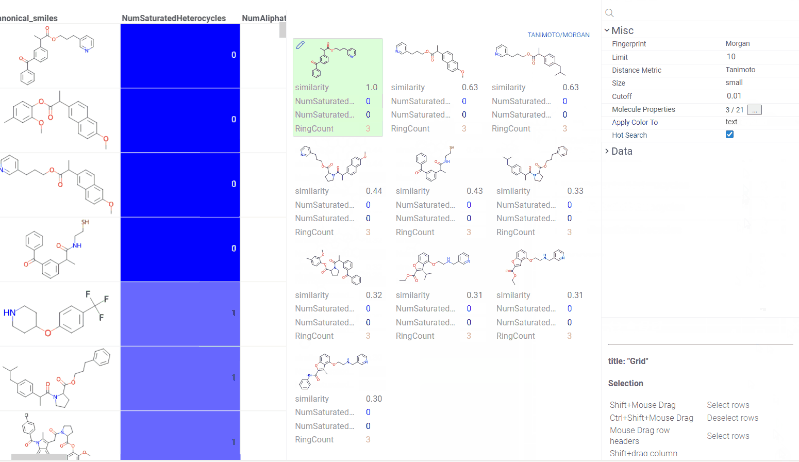
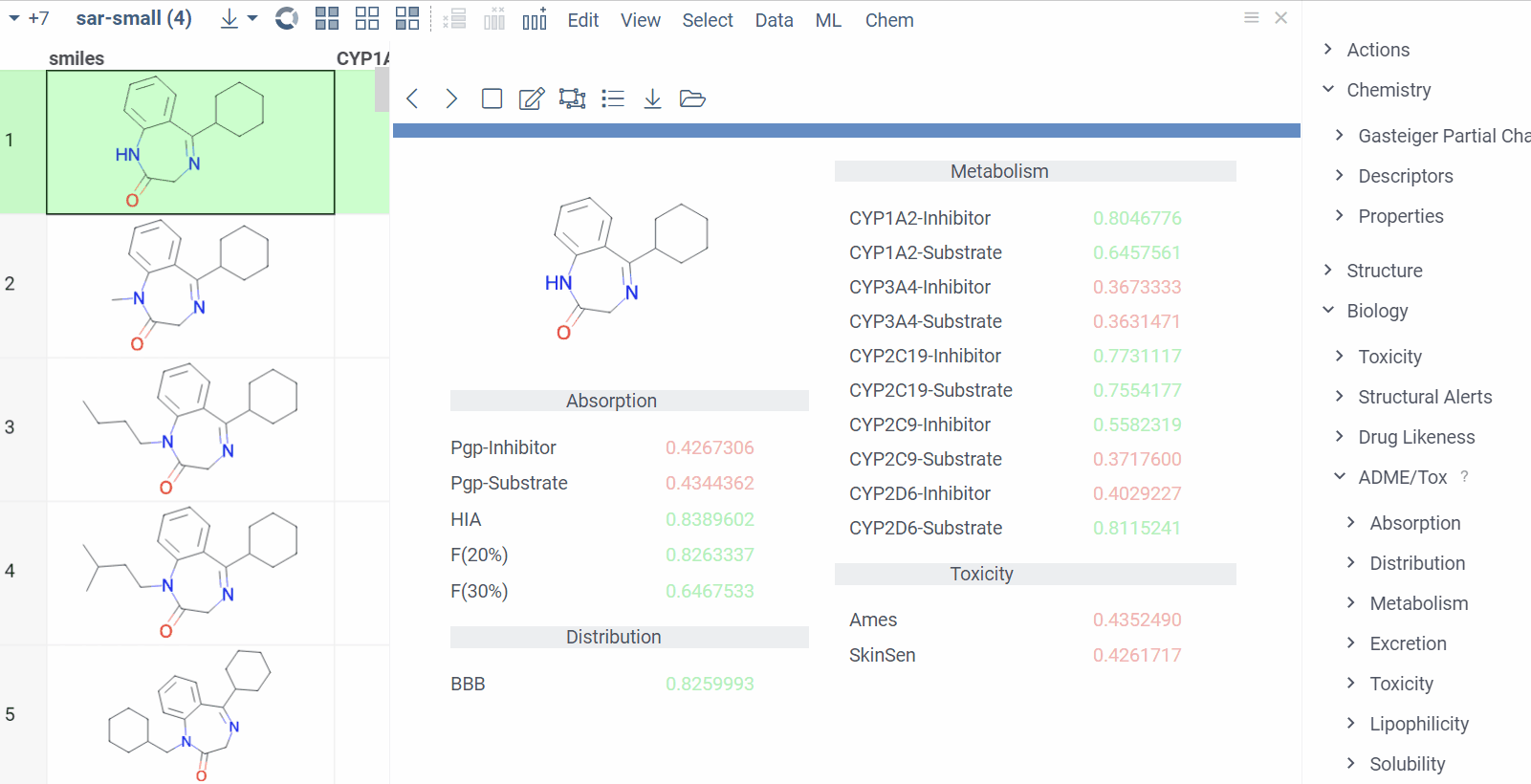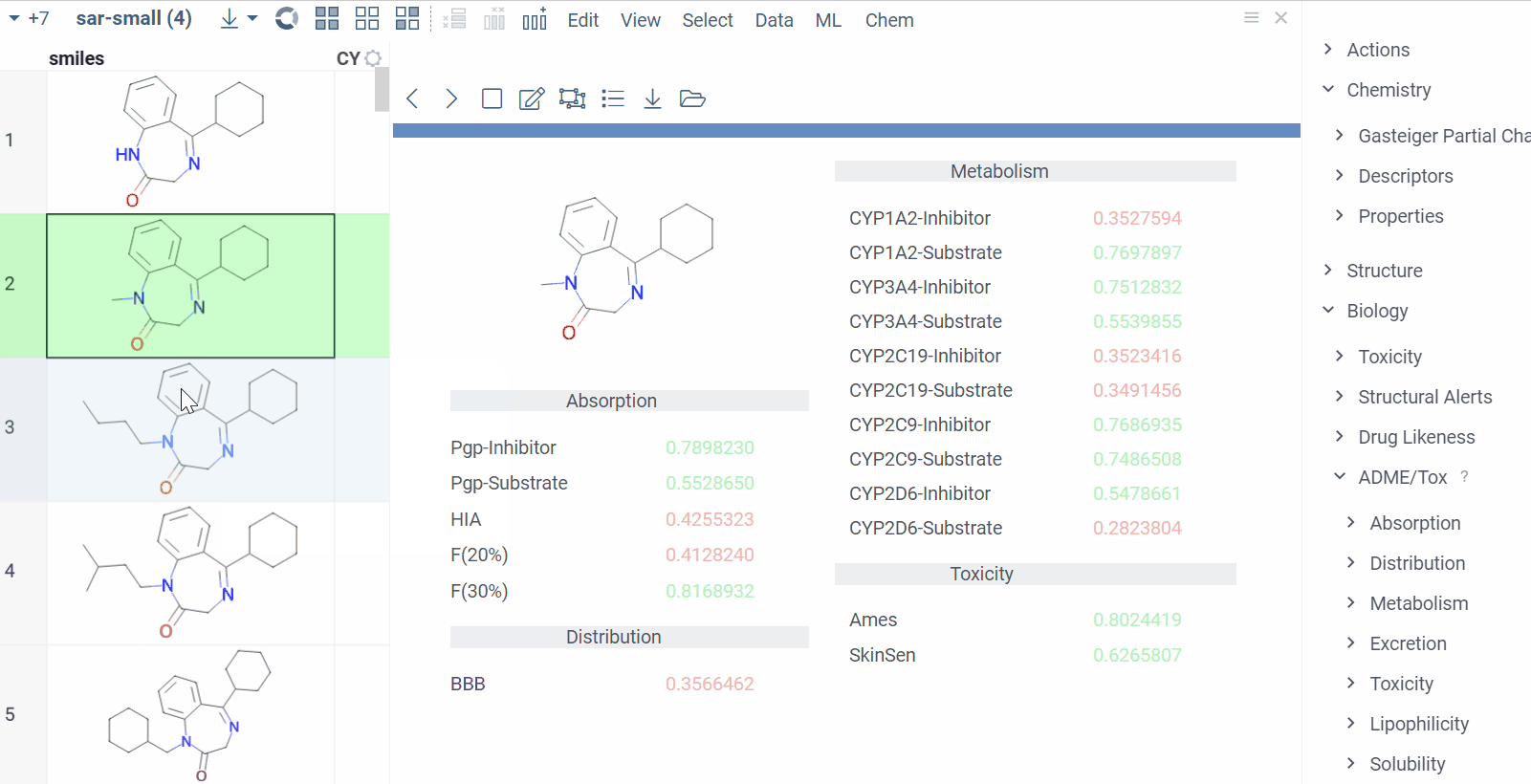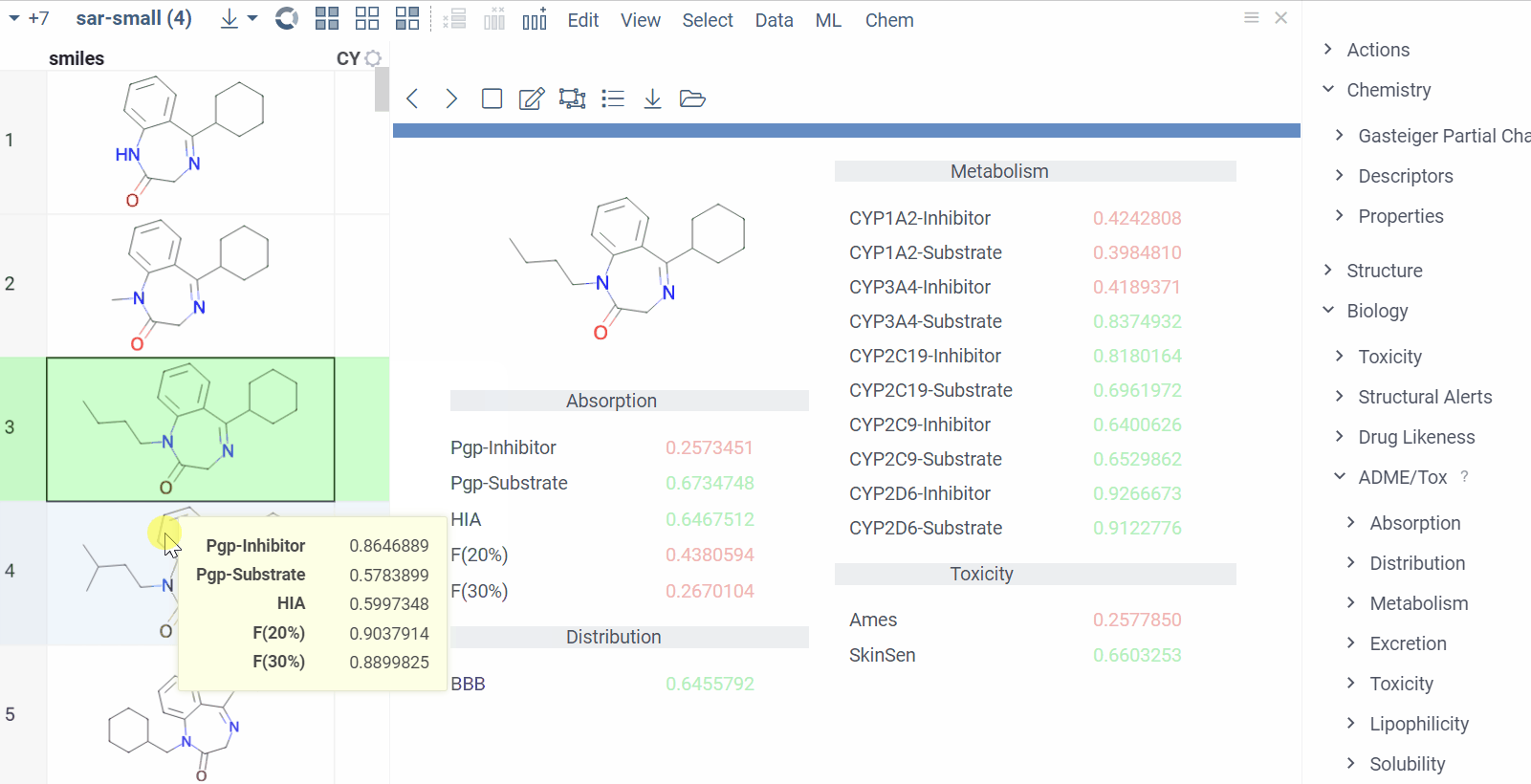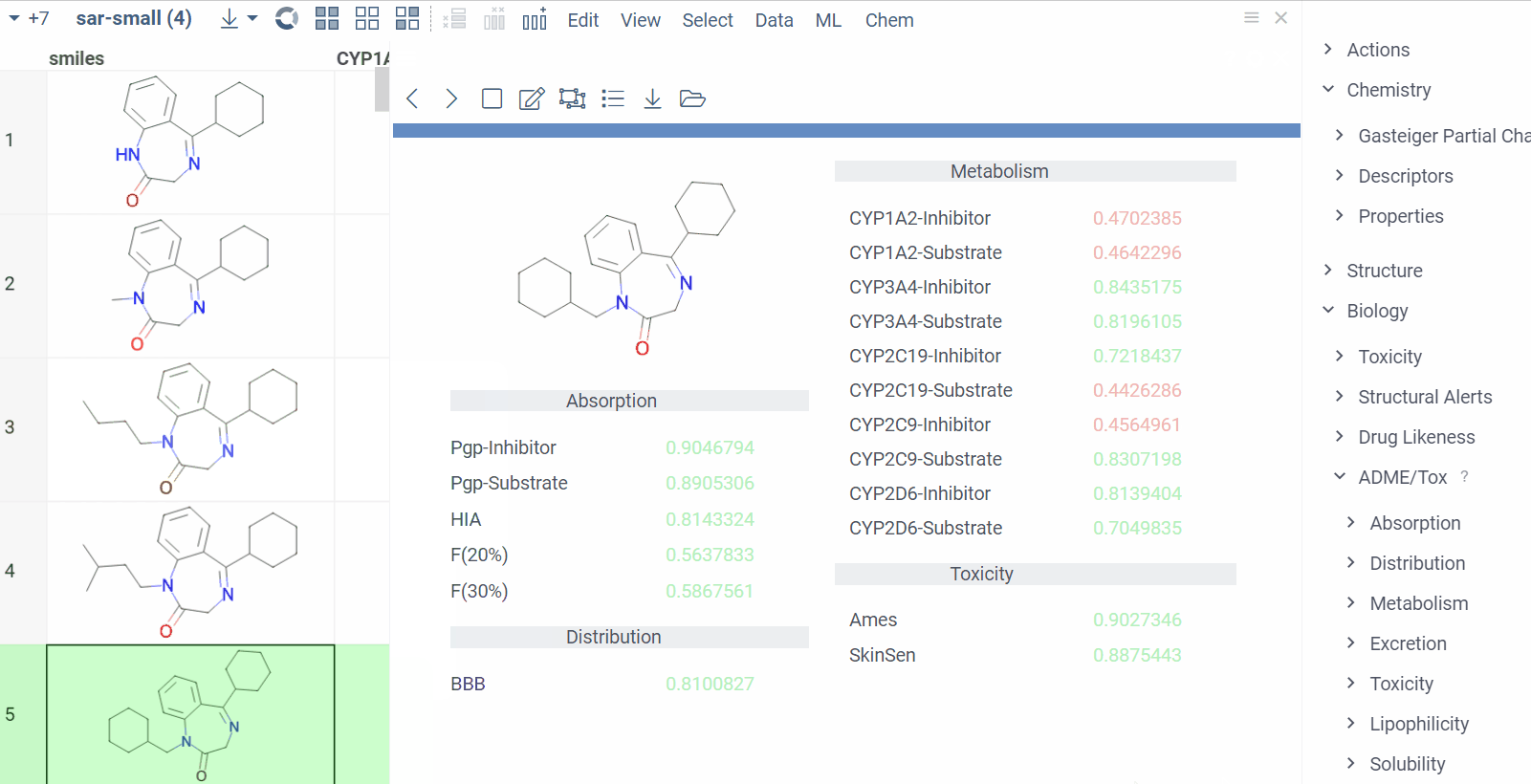
Chem 0.3 (Chem.0.3.0-8b678c) is released, providing for a number of great performance improvements and new functionalities.
-
grok.chem.substructureSearch now provides for both “naive” graph-based search and library-based search via WASM RDKit’s substructLibrary. The latter is chosen as a default for this method. Microbenchmarks are added too (more on their performance is further). Thanks to Paolo Tosco, we are using a pre-release version of WASM RDKit containing substructLibrary methods. This version will soon be merged to RDKit’s MinimalLib master.
-
RDKit-based rendering is now supplied with LRU-caching of RDKit’s molecule structures for both rendered as-is and scaffold-aligned renders. The Coordgen-based rendering is turned off by default (until we improve upon this further in Chem), providing for better scrolling experience.
All this is offered completely in the browser, without involving any server-based computations.
One area of particular interest for our users was the performance of substructure search. In fact, already with “naive” graph-based search, where we match the given pattern molecule to every entry of a dictionary in a loop using get_substruct_match, the performance was reasonable on small datasets. Yet, this wasn’t a match for the most typical use case, where many subsequent pattern searches are applied to a single dataset which changes rarely.
That’s were the RDKit’s substructLibrary comes in handy. It first allows building the so-called library, to which we perform further pattern searches against. Let’s look at some of our micro-benchmarking results of the discussed approach. We are taking a large dataset of 40 000+ molecules, and do a search for 3 small patterns written in SMILES: ['c1ccccc1' (Benzene), 'C1CCCCC1' (Cyclohexane), 'CC' (ethane)]. We linearly increase the dataset being sought through from 2’000 to 12’000 molecules and track the performance of these 3 searches altogether, averaged on 10 independent runs. For the input file demo/chem/zbb/99_p3_4.5-6.csv it is in the picture bottom, and for demo/chem/zbb/99_p1_ph6-8.csv it is in the picture top.
You can check these numbers here at Datagrok https://public.datagrok.ai/p/d9f38d00-2e30-11eb-eb2f-cf7849c78d19. We encourage you to try the microbenchmark yourself and get the figures on your computer: https://public.datagrok.ai/js/samples/domains/chem/substructure-search-library.
It’s clear how faster the substructLibrary-based search is compared to a graph-based one. 10x speed-up is a consistent observation on both 99_p3_4.5-6.csv and 99_p1_ph6-8.csv. We also see that the initialization time of the library is linearly dependant on the dataset size being indexed, and is roughly comparable in time to just a single graph-based substructure search.
These numbers are already great, yet there is a good room for improvements in going parallel and enabling library construction using pthreads via WASM. In fact, the infrastructure for that is already in-place, and we are already figuring out the details of making these threads work efficiently on WebAssembly.
Some plans for the future versions of Chem:
- Make a choice of either OCL or RDKit-based rendering as a package option.
- Make a Coordgen-based or RDKit-native rendering as a package option.
- Add caching (precomputed search structure) to similarity scoring in a similar fashion to substructure search.
- Detach computations in all the cheminformatics methods from the main thread via a standalone JS web worker. That should improve the user experience, as the UI won’t be blocked on processing large pieces of data, even if this is blocked for a few seconds.
- Speed up similarity scoring dictionary search dictionary preparation via parallel computation on JS web workers.
- Speed up substructure search library creation via activating pthread-based parallelism already provided in substructLibrary, but for its WASM version which we are using right now.
Please let us know of the other possible improvements to the cheminformatics functionality shipped via the browser, as well as whether you’d want to see more microbenchmarks from the Datagrok team. We are also going to implement an advanced benchmark based on the summary.