Community
Cheminformatics updates
Announcements
chem
skalkin
December 3, 2022, 8:22pm
18
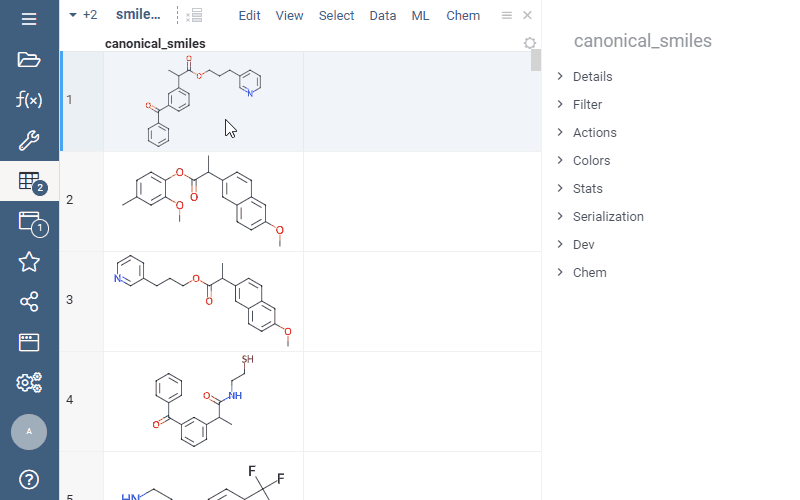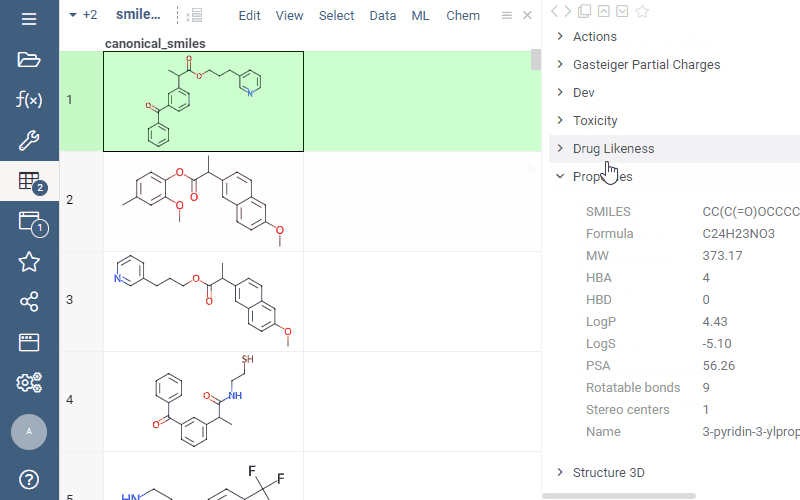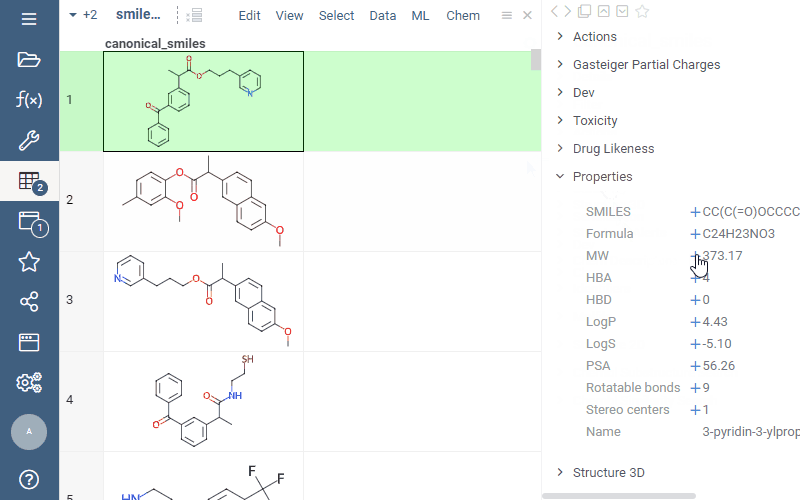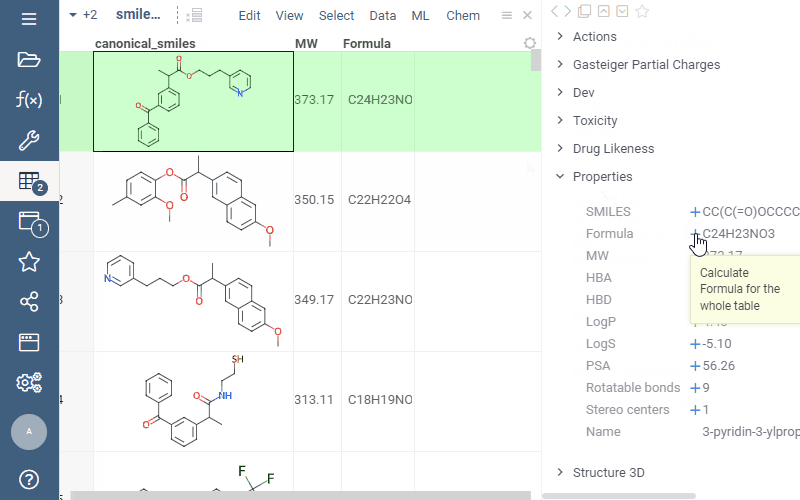
Now you can easily add calculated properties right from the “Properties” panel:
molecule-properties
800×500 161 KB
1 Like
Where is cLogP?
show post in topic