In which version of the Chem Package is ADMET included?
Hi Tom, apologies, it looks Oleksandra pulled the trigger a few days too soon ![]() AdmeTox is a new plugin that will be released in a couple of days.
AdmeTox is a new plugin that will be released in a couple of days.
1 Like
Hello, is the package already available?
I’m using Chem version 1.6.15 but I can’t see ADME/TOX menue in Chem tab.
Thanks,
Hello,
AdmeTox is a standalone package located in the Uncategorized section (Manage > Packages > Uncategorized). Currently, version 0.0.1 of AdmeTox is available.
We sincerely apologize for the inconvenience caused by the unavailability of the package you were expecting. Datagrok team is currently working on the remaining technical issues that are crucial for the successful release of the package. Our main focus is to ensure its stability and meet all the necessary requirements, which will ultimately enhance your experience with the product.
We deeply regret providing incorrect information about the package being available when it was not. It was an oversight on our part, and we understand the confusion it caused.
Please be assured that we will test the package and release its new version as soon as possible and will promptly update you with the revised information.
2 Likes
New substructure search functionality is available in the Chem package version 1.7.0.
We drastically improved the algorithm, and now it provides the following:
-
ability to filter datasets containing millions of molecules without memory overflow
-
10 times faster consecutive searches against the same dataset. This is done by caching and reusing the pattern fingerprints
-
The first search results appear in just a split second, regardless of the dataset size. You do not have to wait for the entire dataset to be filtered before exploring the results.
-
Easy search process tracking with a search progress bar
There are two ways to run a substructure search:
- Select
Top menu→Chem→Search→Substructure Search... - Click Filter icon on a toolbox
In both cases, a filter opens on the right side of the table.
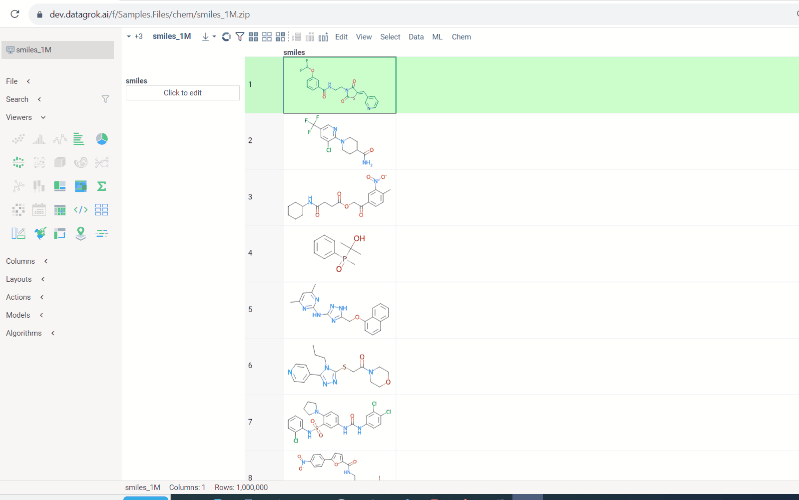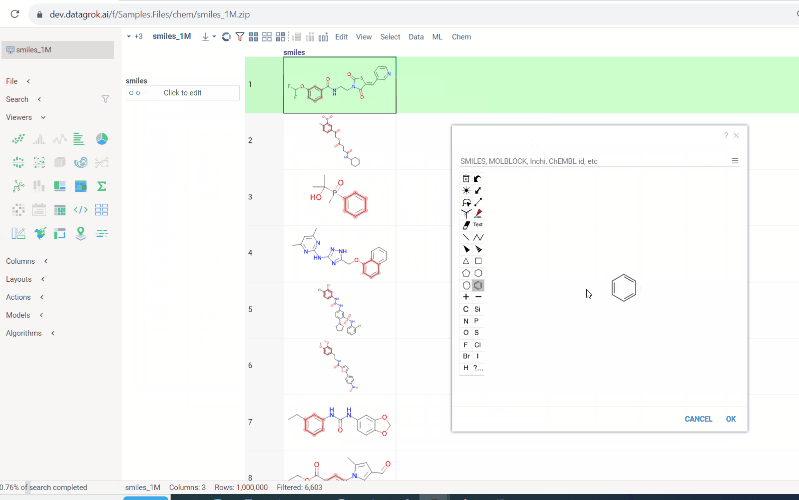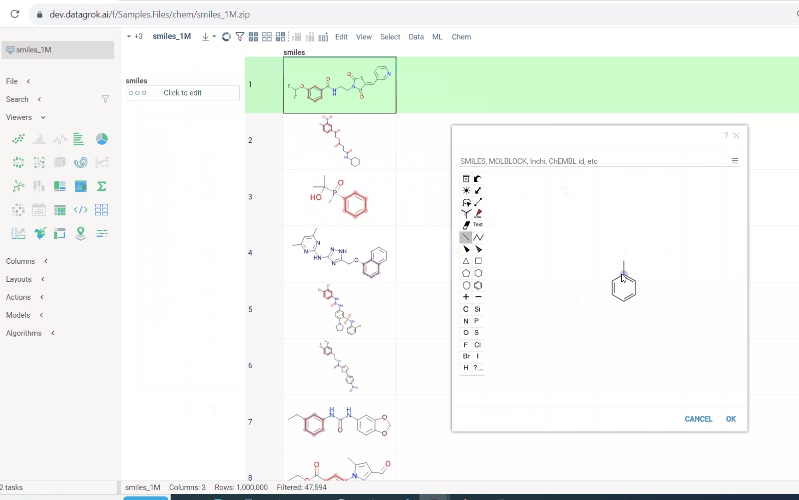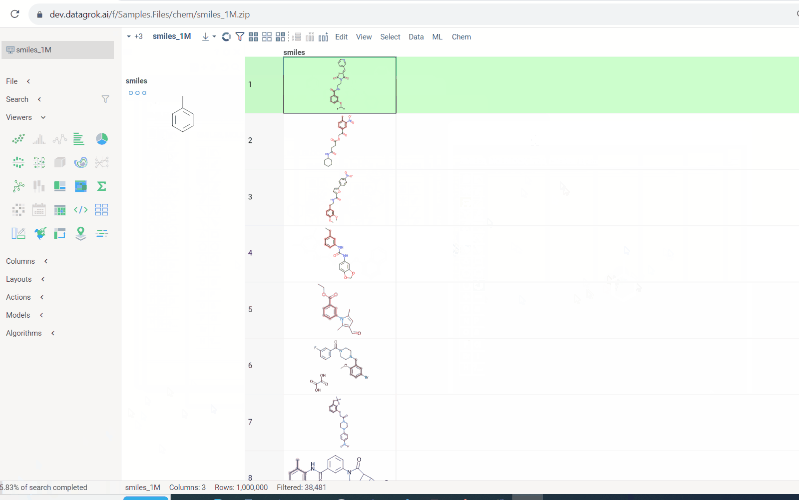
To conduct a search:
-
Click on the
Click to editpane for the desired column. The molecule sketcher opens -
Sketch some substructure. The search starts immediately.
Clicking OK during the active search closes the sketcher, but the search continues. Clicking Cancel terminates the active search.
Additionally, you can modify the substructure during an active search, and the results will recalculate instantly.
Here is an example:
2 Likes
Hierarchical clustering for Molecules
Hierarchical clustering function from Dendrogram package now supports Molecule columns in various formats.
Users can also cluster Molecule columns alongside with numeric or Macromolecule(sequence) columns. In such cases, Distance matrices will be calculated for both, normalized and added together.
To run hierarchical clustering, Select Top menu → Chem → Analyze → Hierarchical Clustering...
Then, select the column with molecules, choose the linkage method and press Ok.
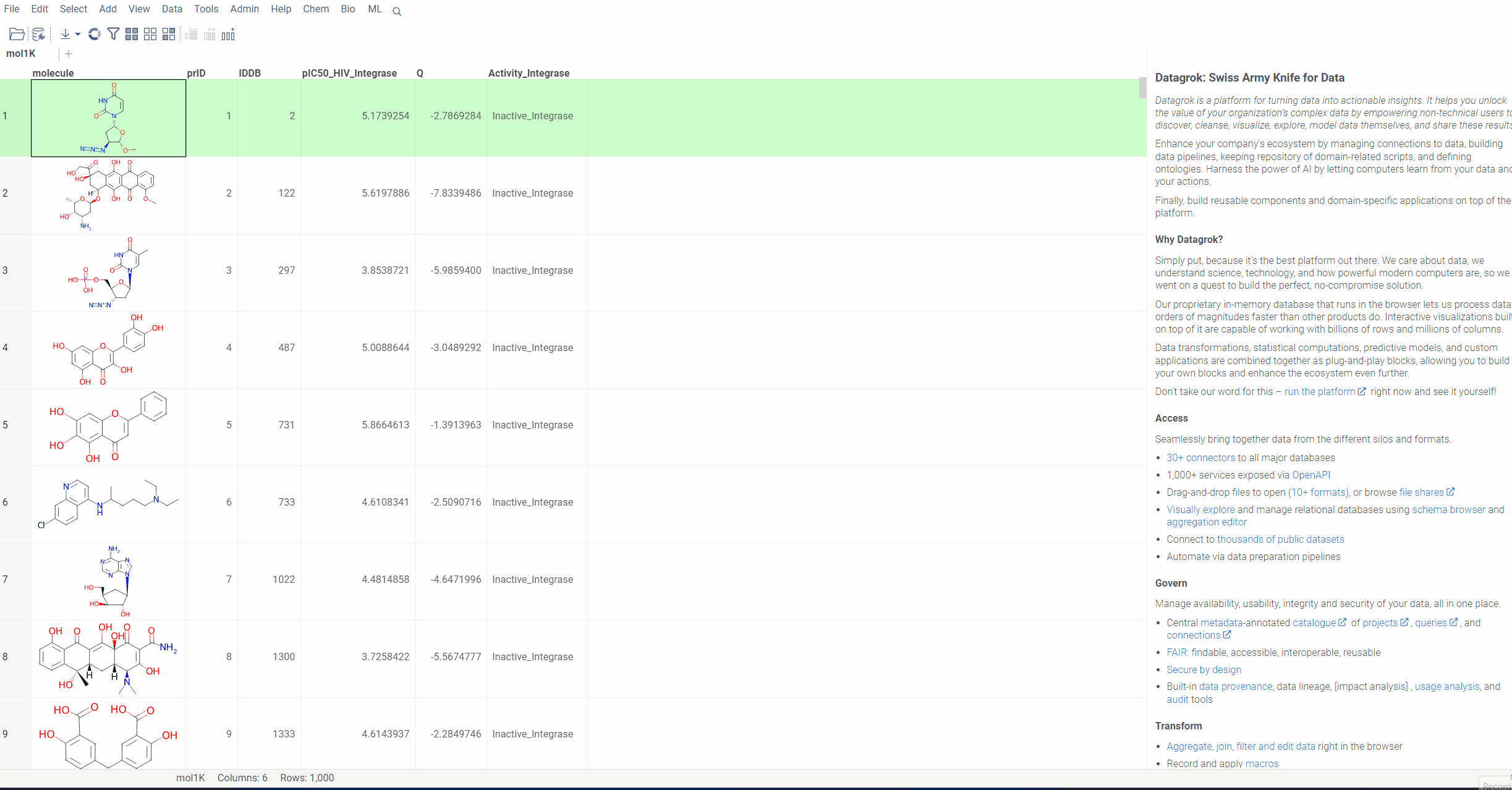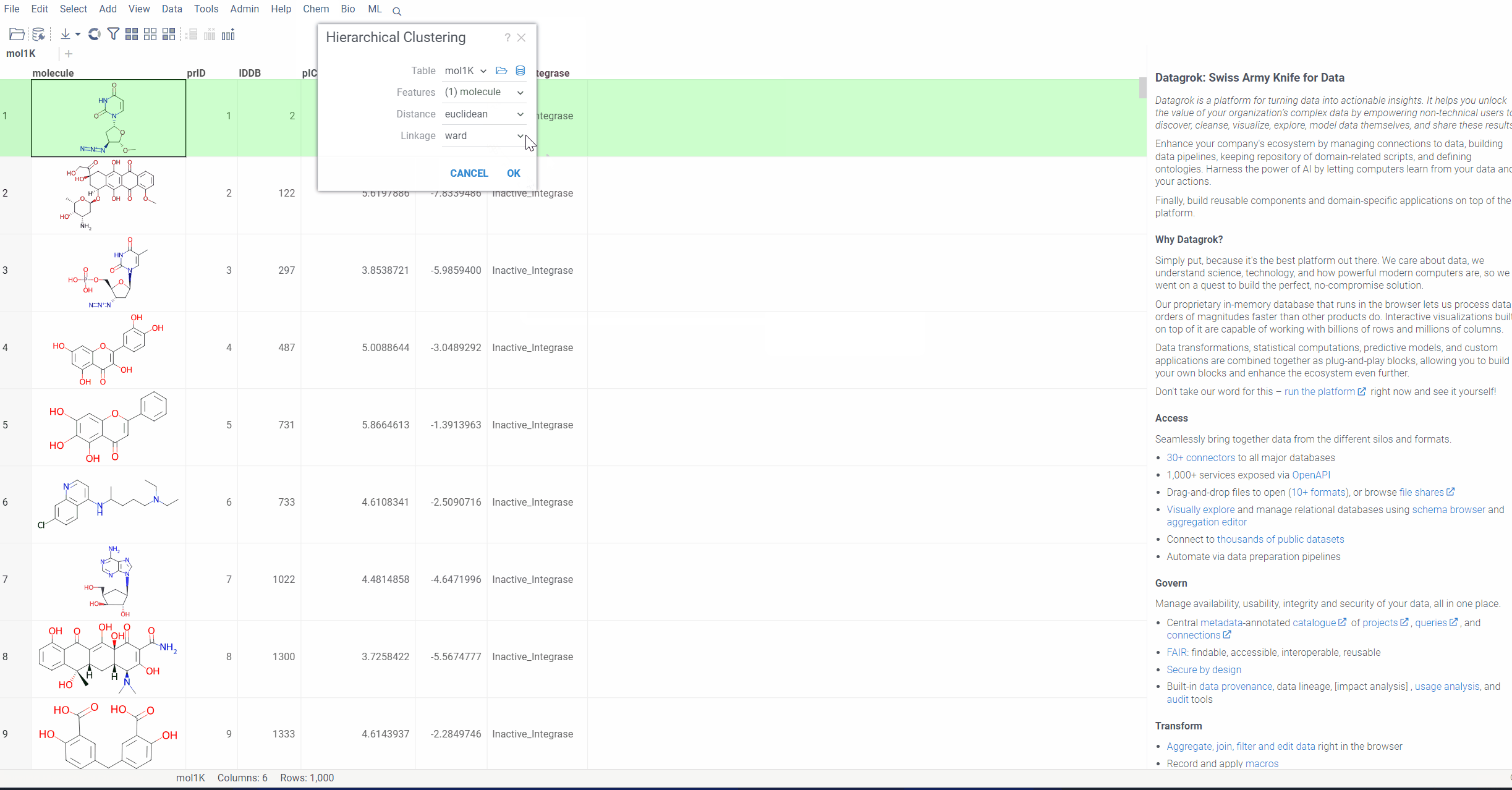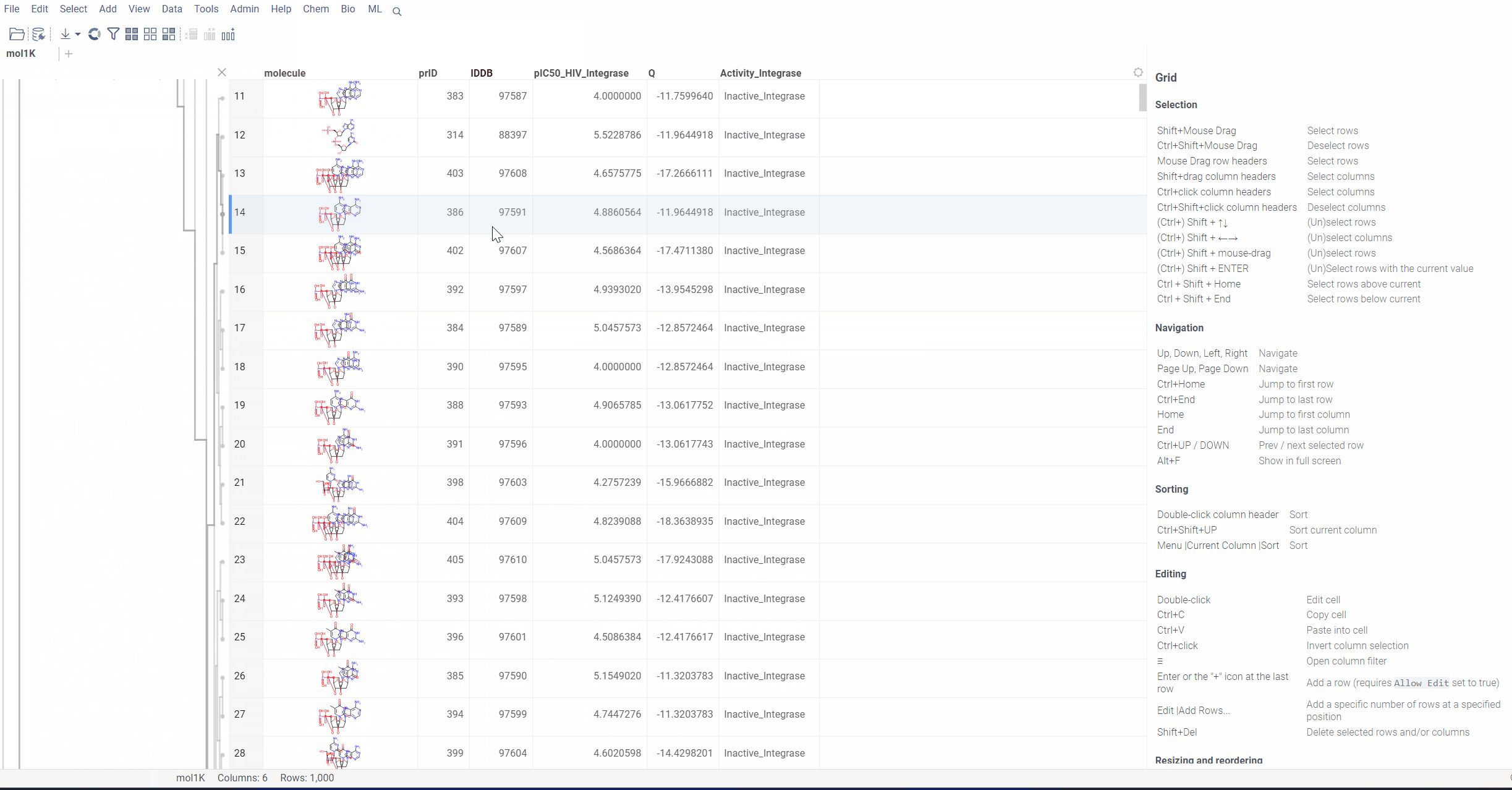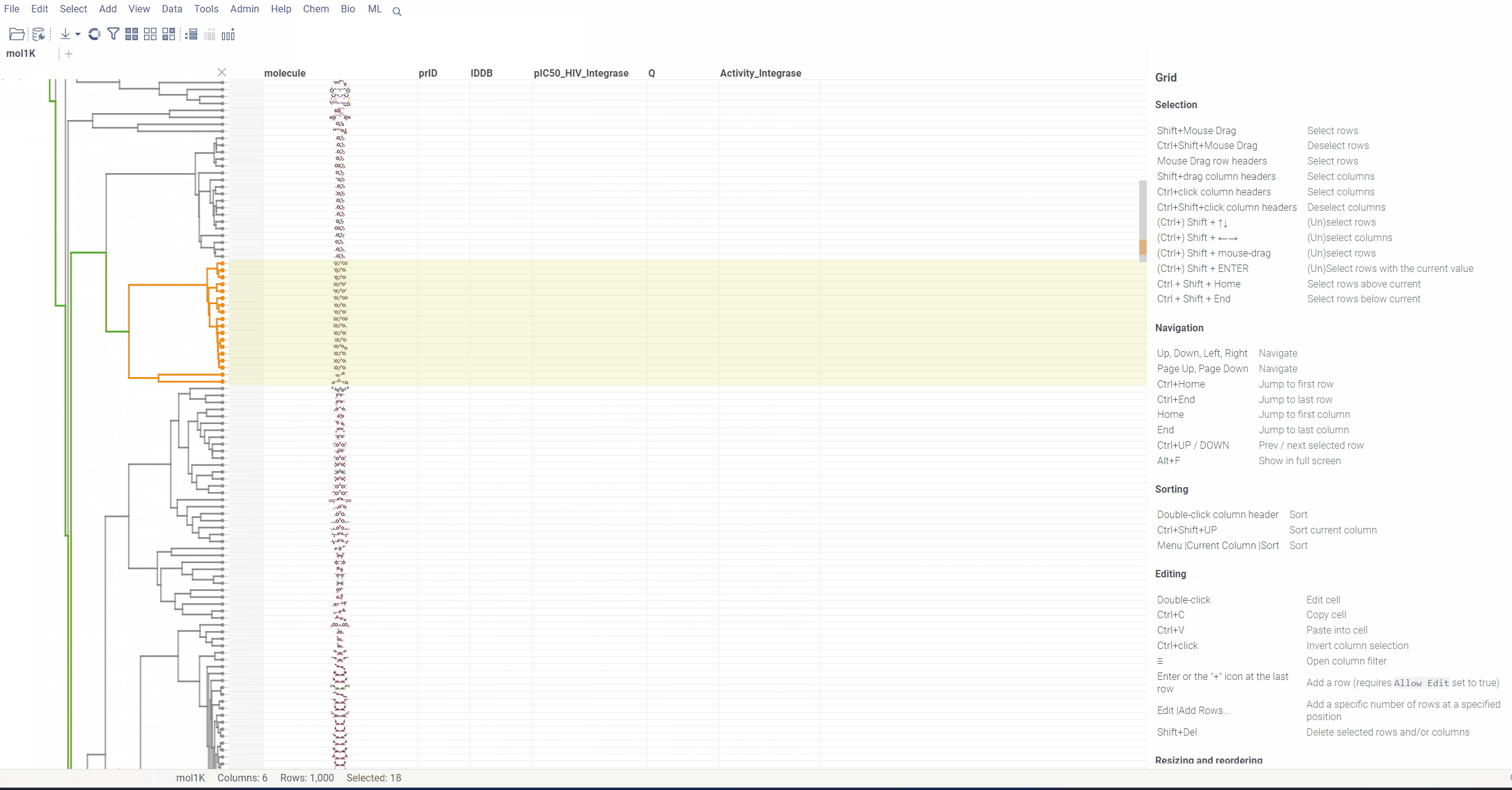
Distance function:
First, for all molecules in the column, we calculate ‘Morgan Fingerprints’, which are 2048 bit vectors.
For distance function, we compare them using ‘Tanimoto similarity’ metric and build a distance matrix on each pair. This distance matrix then passes on to web assembly code for clustering. All this makes the process incredibly efficient and fast, taking less than 2 seconds on 1000 molecules.
2 Likes
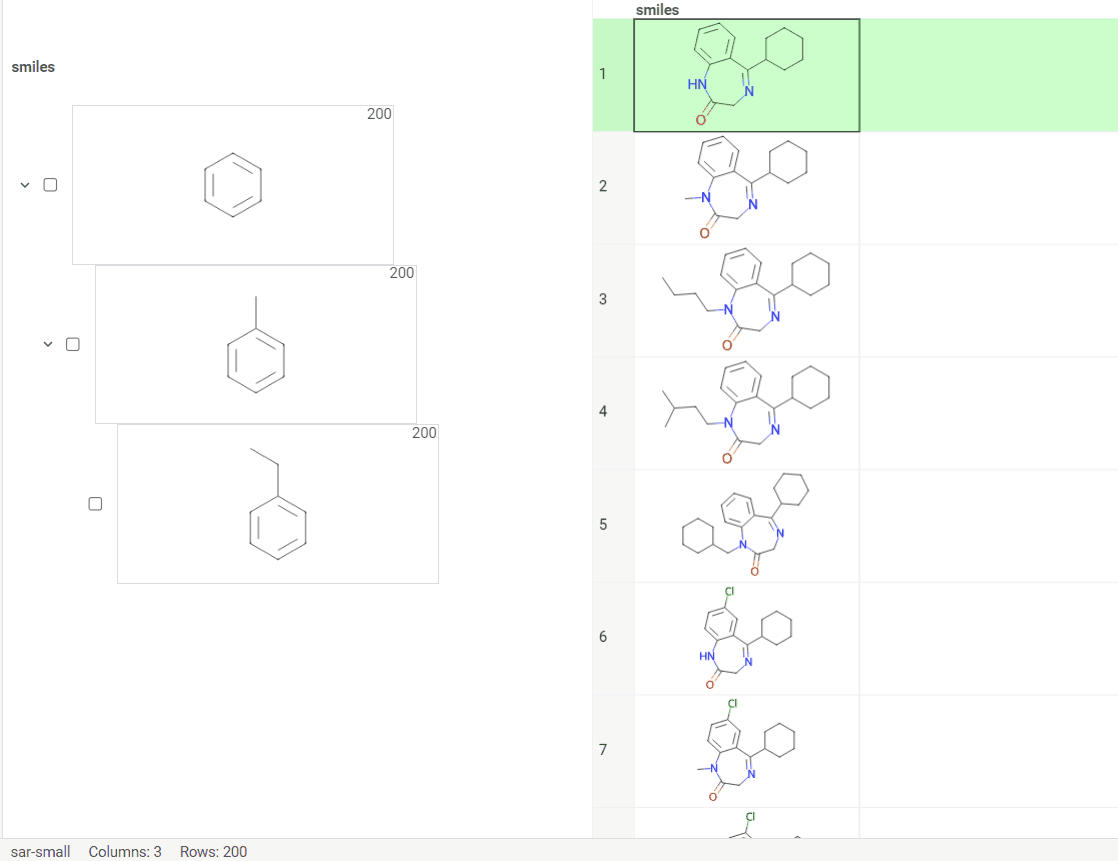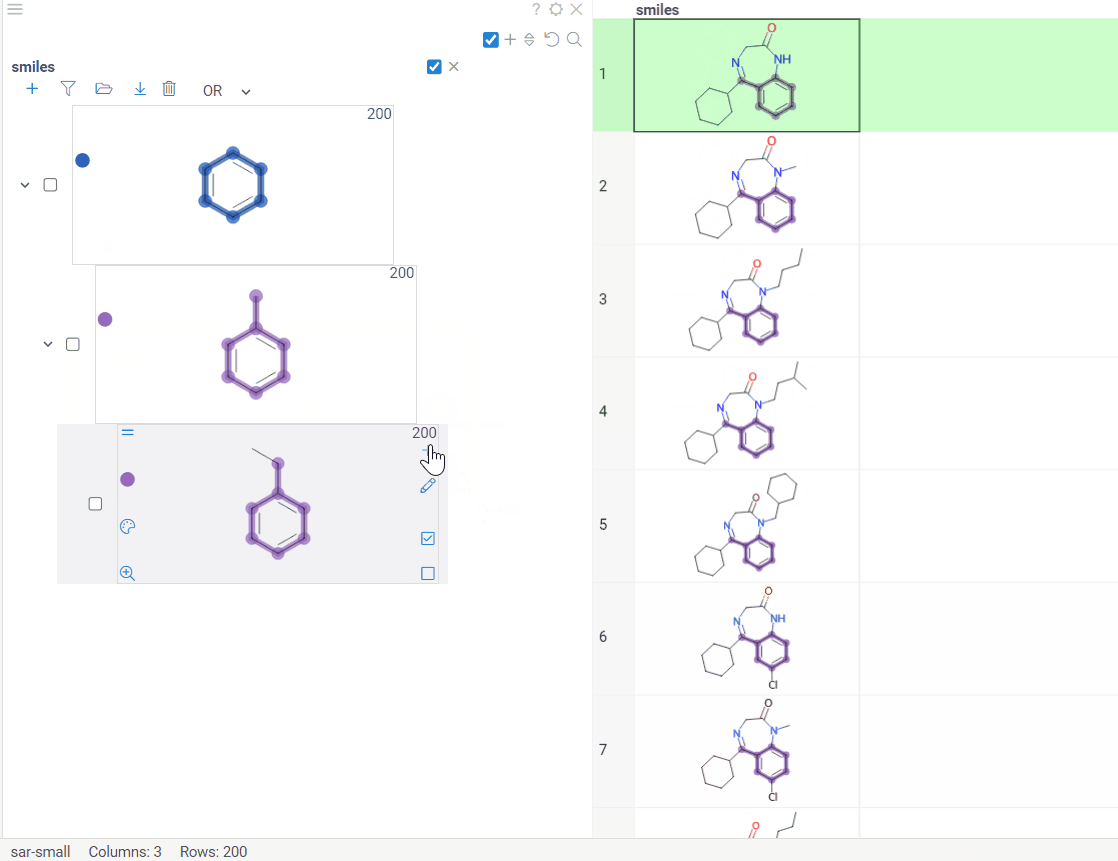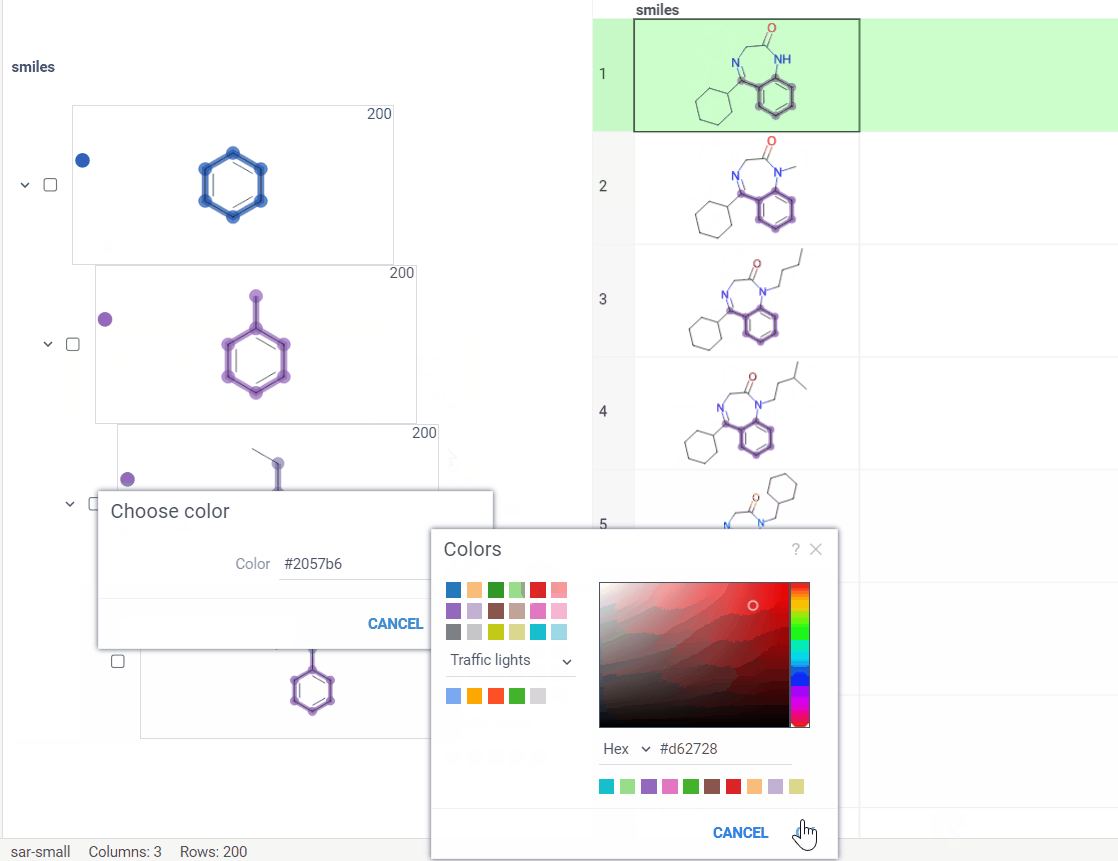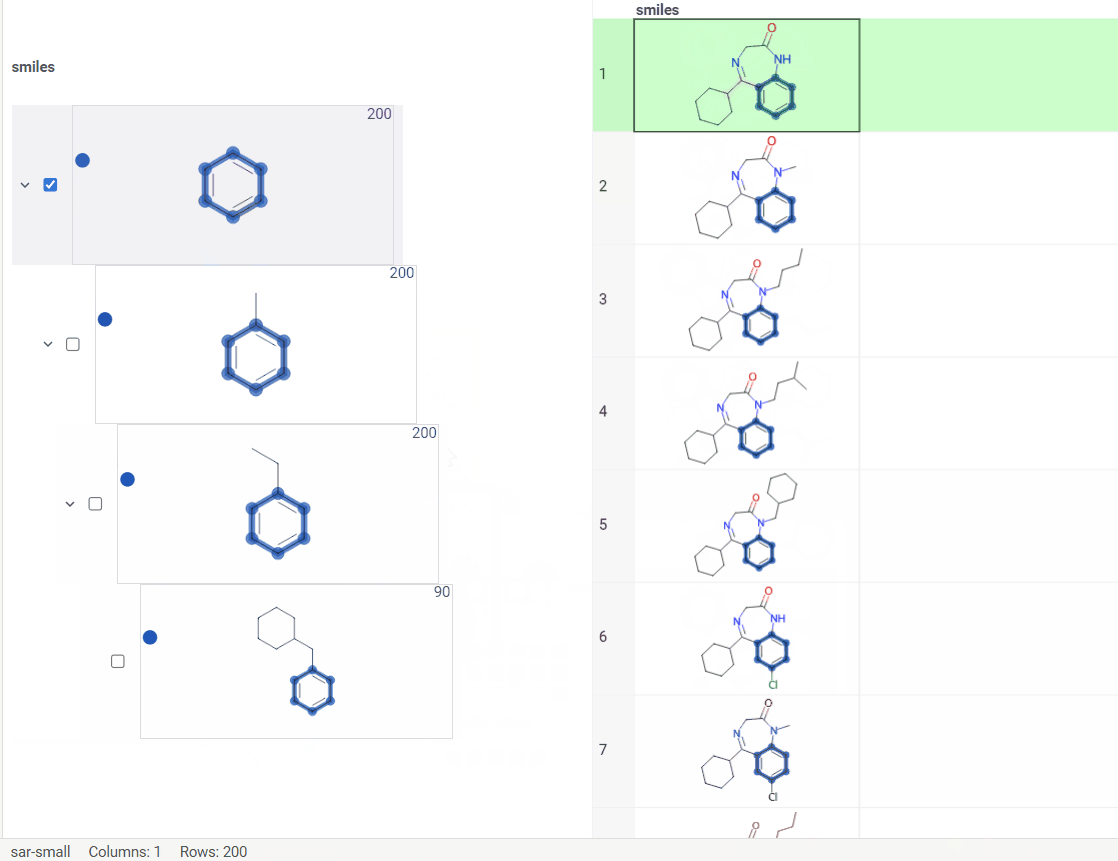
We are thrilled to announce our latest update, Scaffold Tree integration with molecule fragments highlighting. This enhancement makes your data visualization experience even more user-friendly and intuitive.
To utilize this feature, we have introduced two icons:
-
Circle icon. Click to toggle the color on and off. -
Palette icon. Click to open a convenient dialog with color picker. The icon allows you to apply or modify colors effortlessly.
In addition, we have outlined some important guidelines for you:
-
Color propagation. When you select or modify a color, it automatically extends to all child nodes. Keep in mind that more specific structures, closer to the leaves, take precedence. -
Color inheritance. When you turn the color off, the node inherits its color from the parent.
Scaffolds are highlighted both in the column and viewer for the convenience.
For further information, please follow the link.
We believe, that with these enhancements, Scaffold Tree becomes an even more powerful tool for visualizing your data with precision and ease. Explore the new features and elevate your data visualization experience today!
2 Likes
New types of molecular search
Now we support multiple types of molecular search, which are:
- Contains (search by substructure)
- Not contains
- Included in (search by superstructure)
- Not included in
- Exact (search by exact structure match)
- Similar (search by similarity score)
To run the search:
- Click Filter icon on a toolbox. The filter panel opens. The default search type is
Contains. Change the search type by selecting from a dropdown list. - Click
Click to editpane for the desired column. The molecule sketcher opens - Sketch some substructure. The search starts immediately.
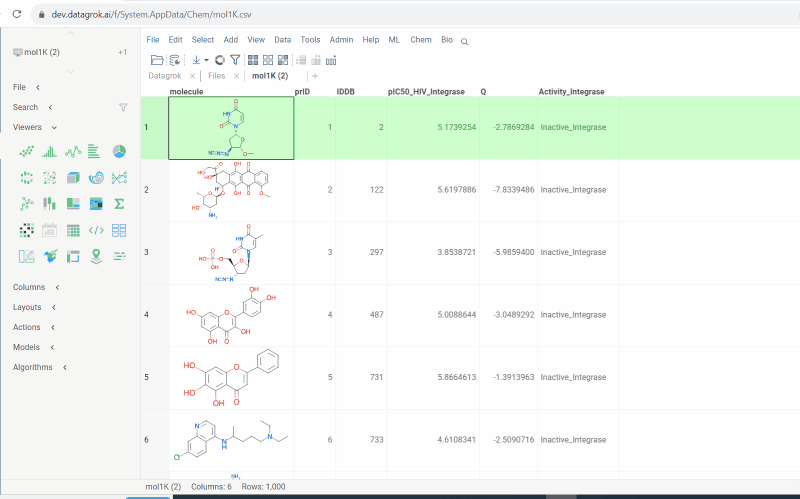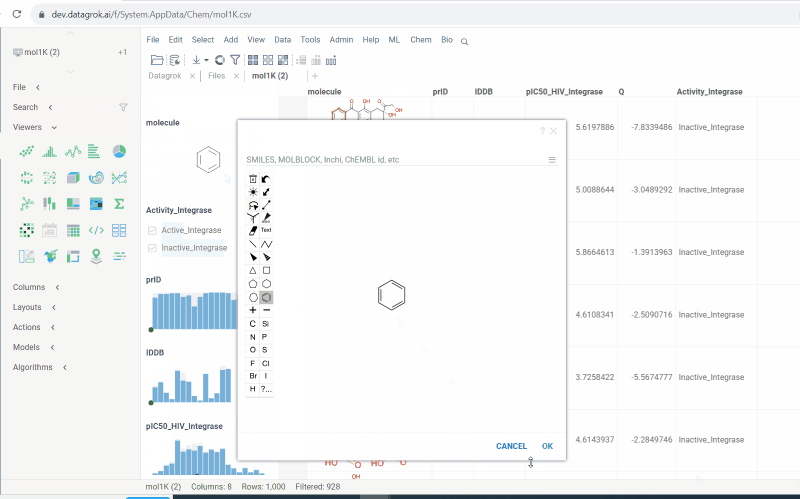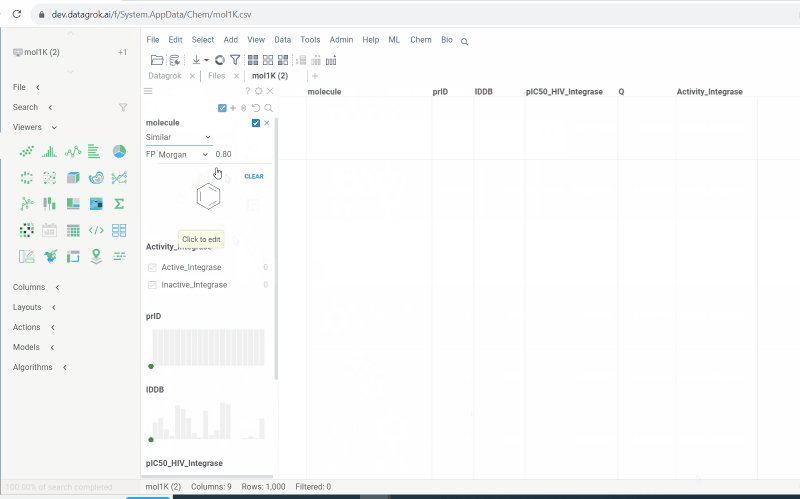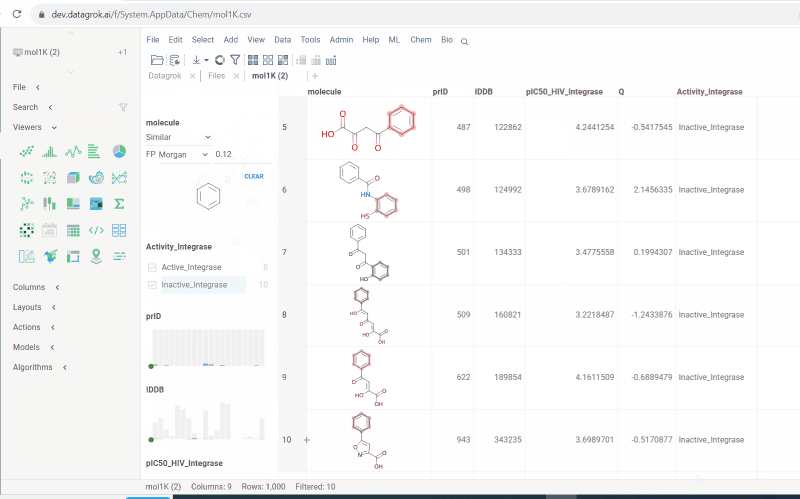
This functionality is available in Chem package starting from version 1.8.0.
2 Likes
Multiple molecule fragments highlight
Now we offer highlighting of multiple molecule fragments inside one molecule structure. Multiple fragments can be highlighted with different colors.
To add a fragment use:
- Context panel
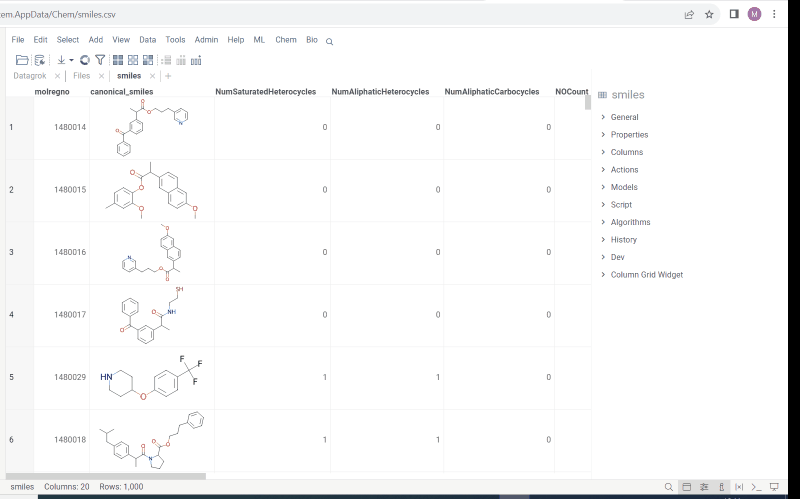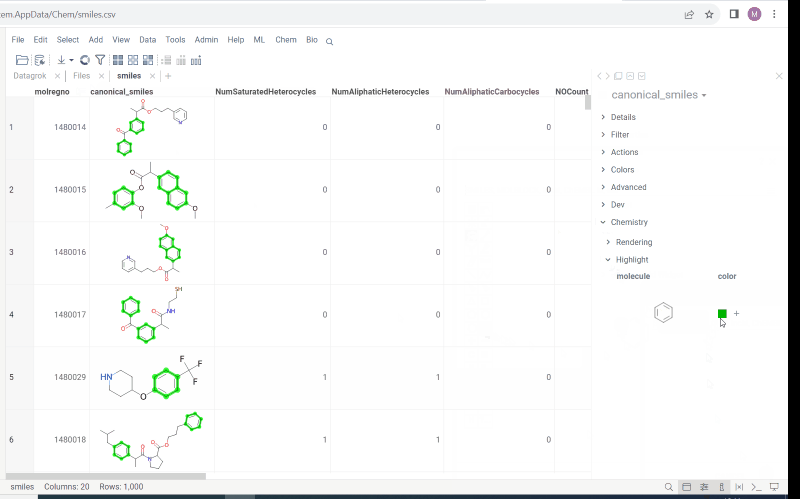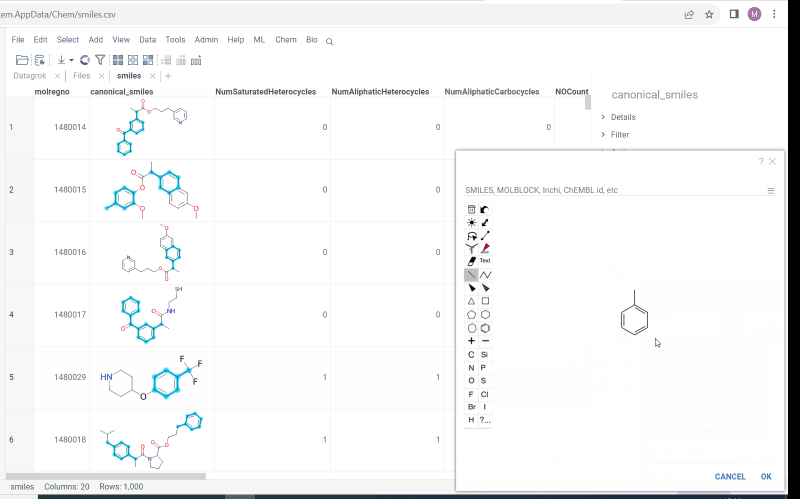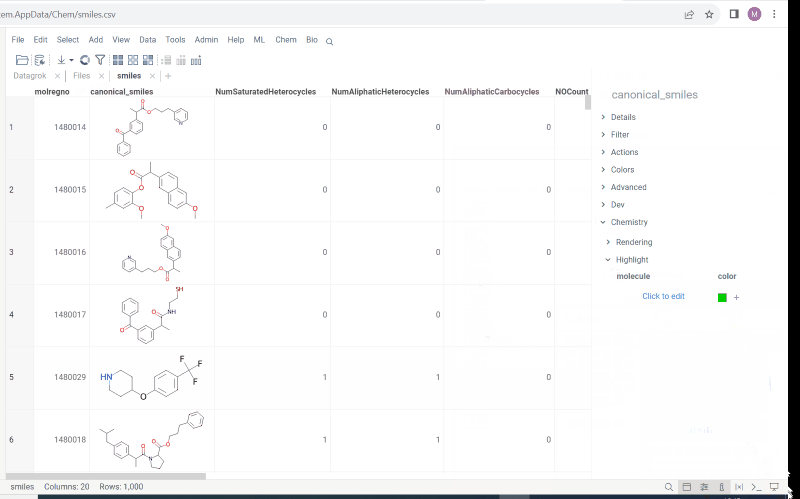
- Structural alerts widget (this feature allows to easily monitor your dataset for multiple structural alerts)
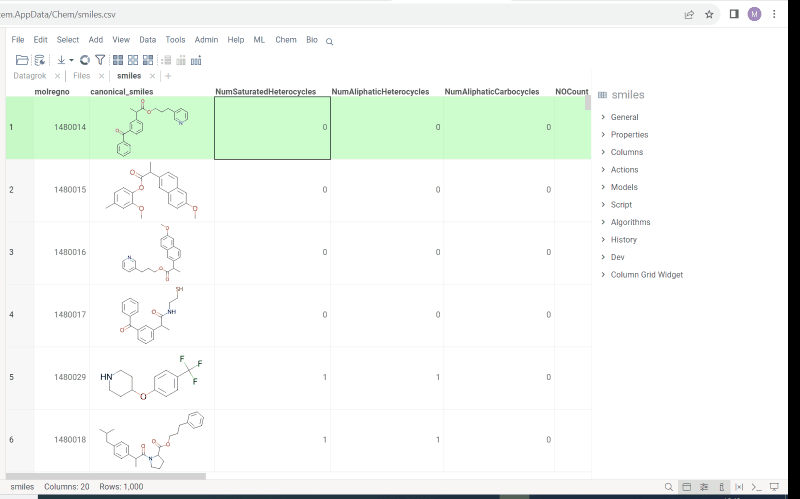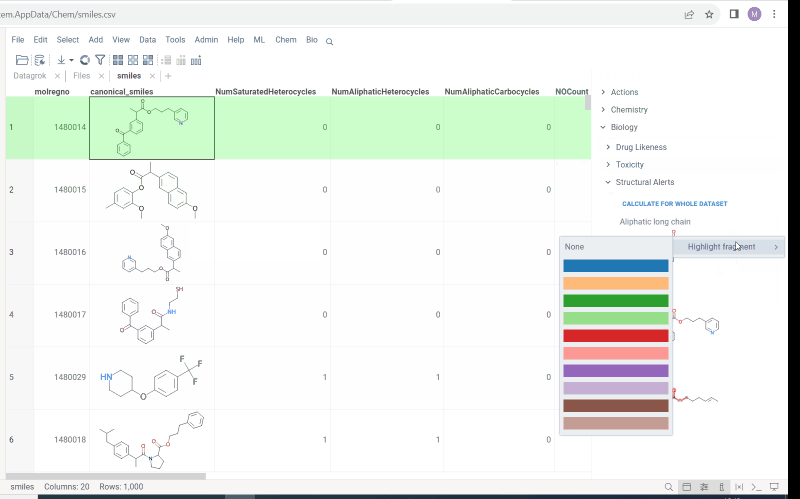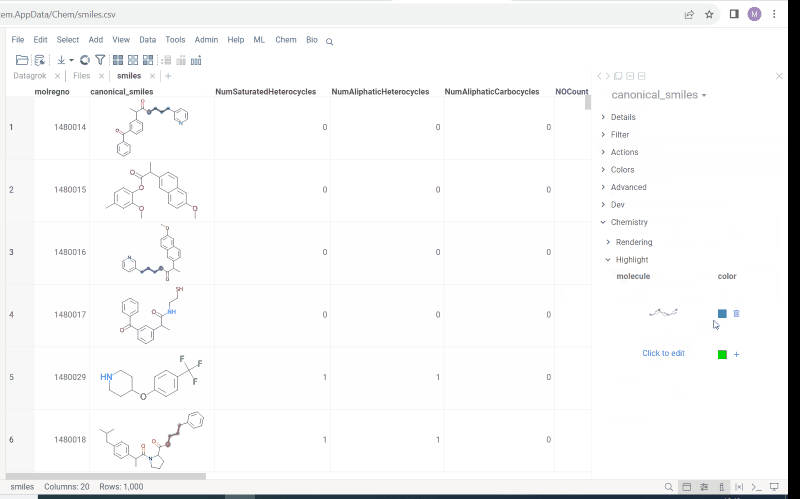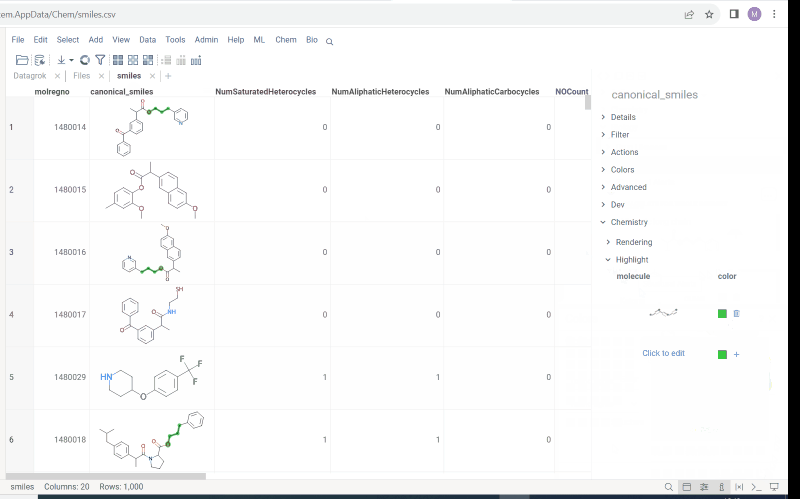
For more information follow the link.
This functionality is available in Chem package starting from version 1.8.0.
2 Likes
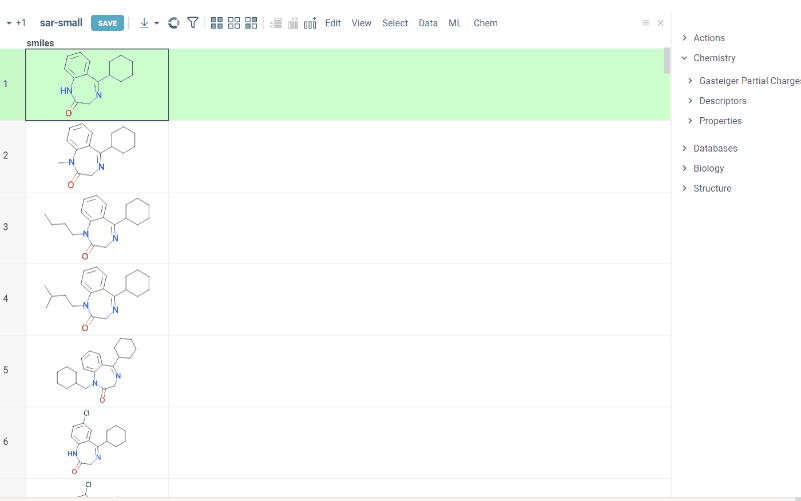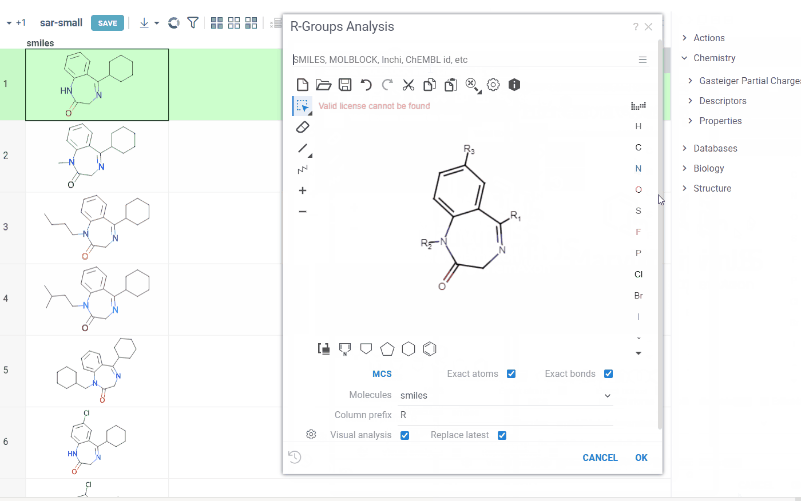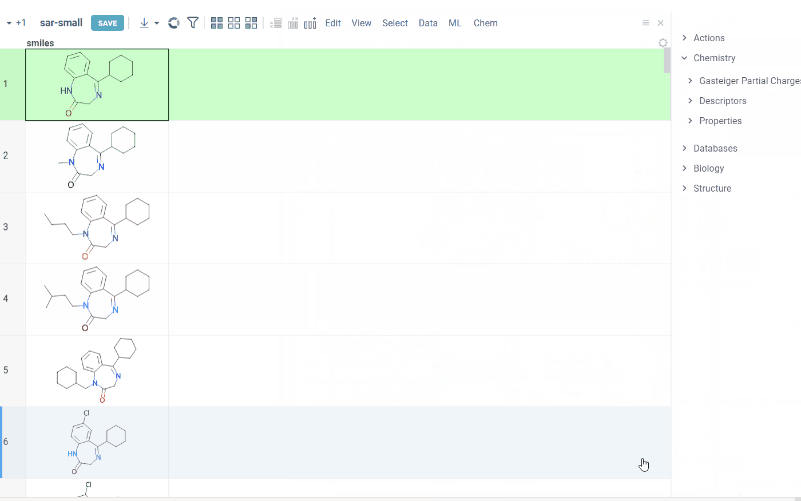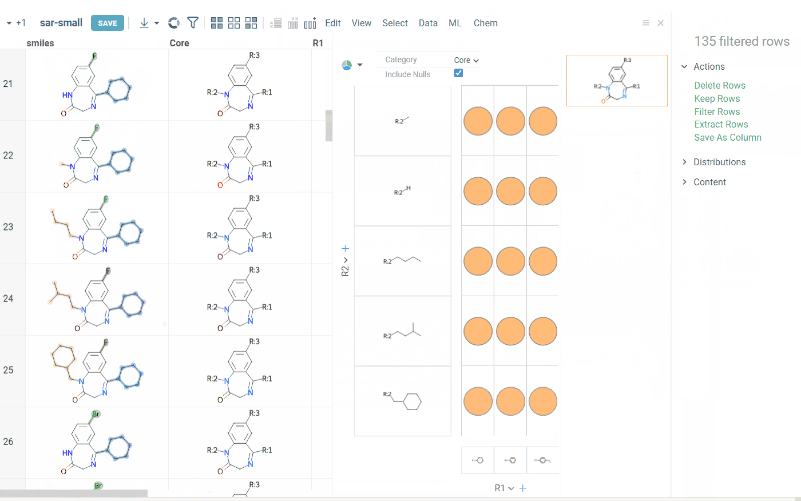
We are happy to introduce new version of R group analysis, which became significantly faster due to running directly in browser with RDKit JS library. New R Group analysis is available in Chem package version 1.9.1
Also there are some extra features available:
- You can define core with custom enumerated R groups
- R groups are highlighted in the initial molecule
- Initial molecules are automatically aligned by core
- You can filter molecules with R groups in each enumerated position using isHeat column
- Additional analysis parameters:
a. Use Replace latest to remove previous analysis results when running the new one
b. Click the Gear icon to adjust R group analysis settings
1 Like
You can now recalculate similarity/diversity search on filtering using Recalculate On Filter property.
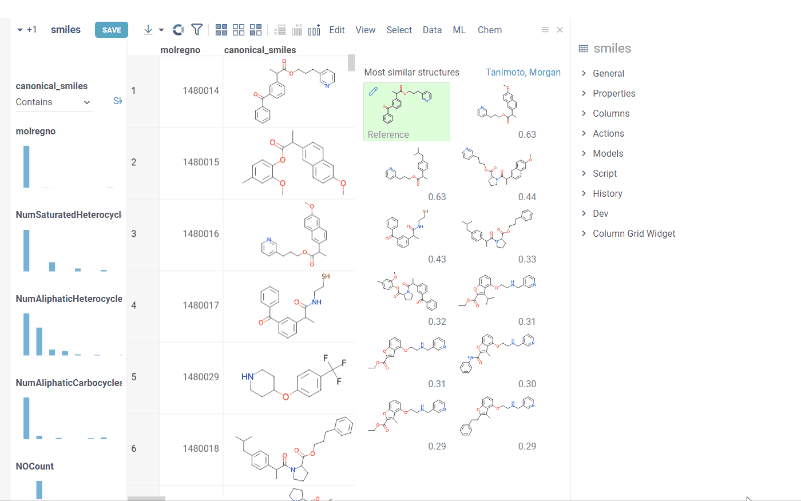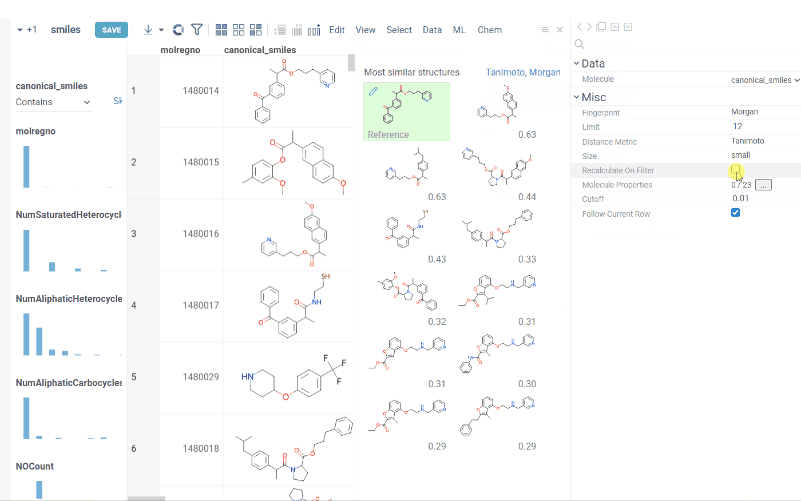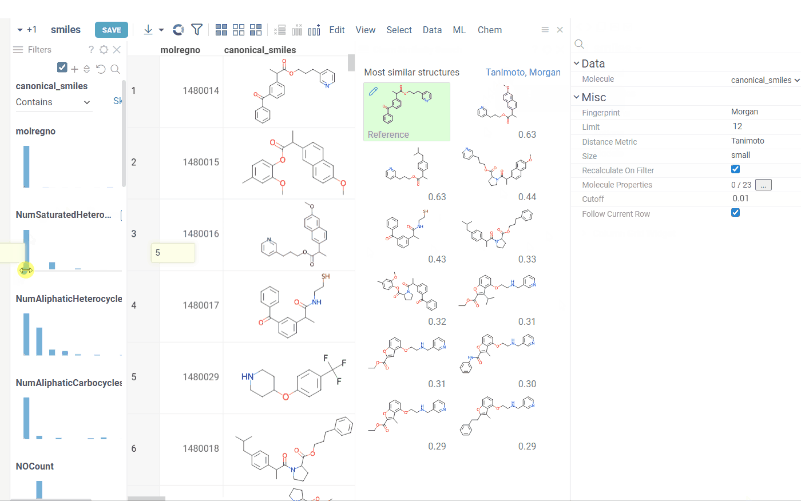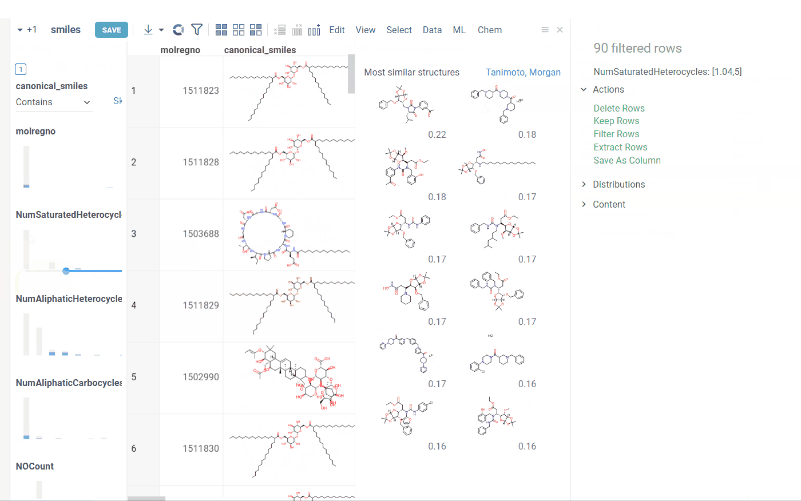
1 Like
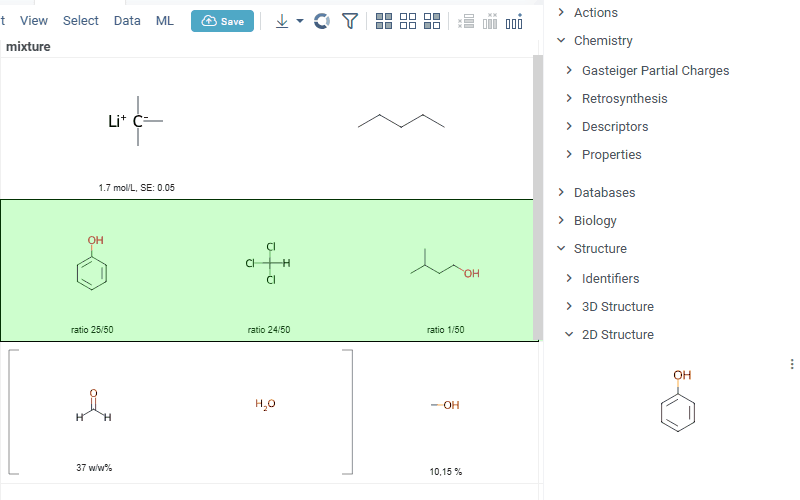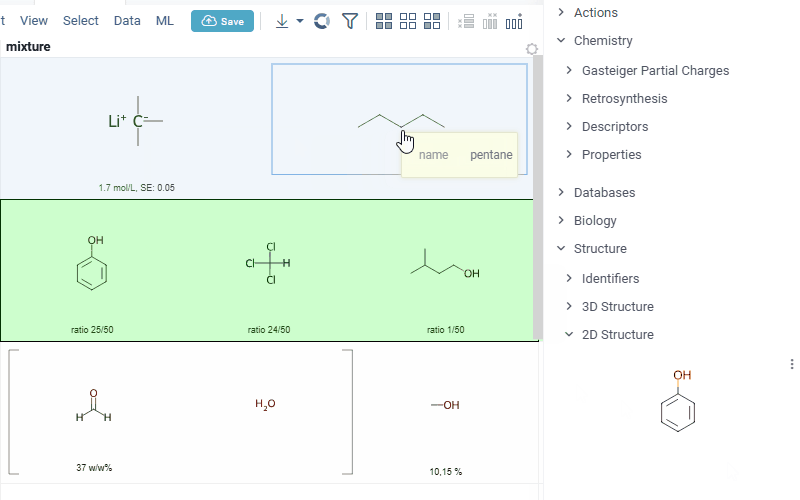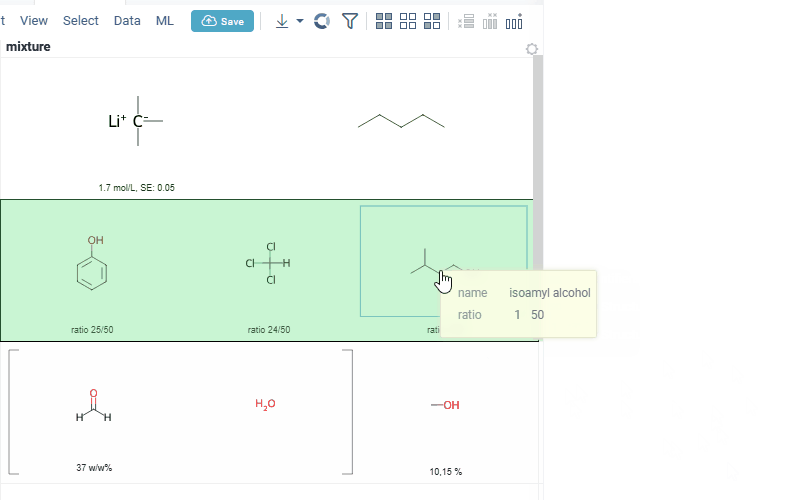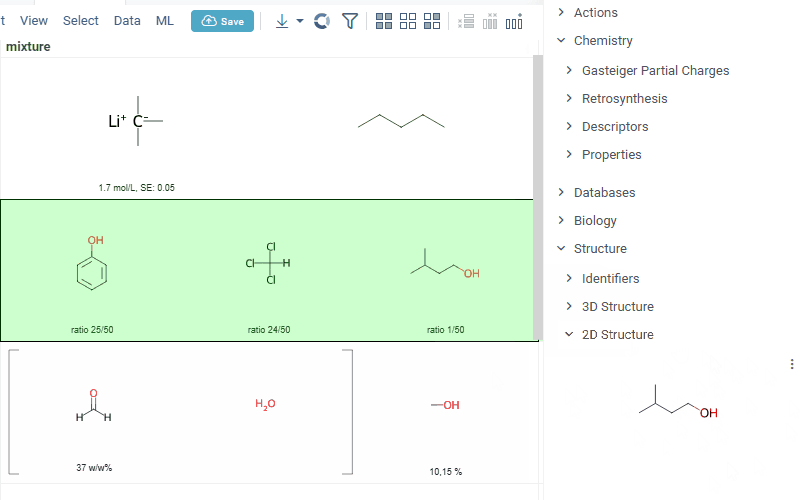
Datagrok is now capable of detecting and rendering mixtures. Mixtures should be in a Mixfile format introduced by Collaborative Drug Discovery, Inc. This fromat is a well-structured json containing comprehensive information about the mixture and its separate components.
Mixfiles are automatically detected by Datagrok and the data is assigned with ChemicalMixture semantic type, allowing to render mixtures of any complexity and retrieve detailed information through the context panel.
The main features are:
-
rendering of the whole list of components in one cell with clear distinguish between components (square brackets are used for nested mixtures). In case component quantity/ratio is present in the Mixfile, it is also shown within the cell
-
interaction with each mixture component straight from the grid cell. Hover over any component to get more details in the tooltip. Click on separate component to get structure details in the context panel
-
summary mixture information in the context panel. You can browse the mixture using either table or tree view
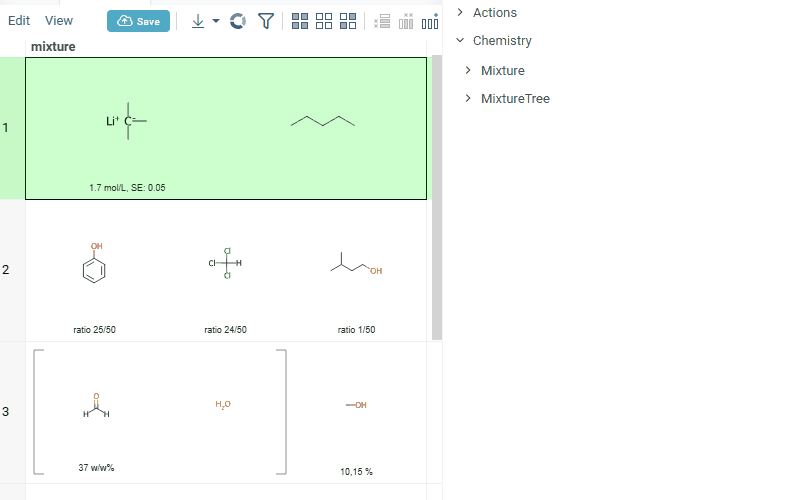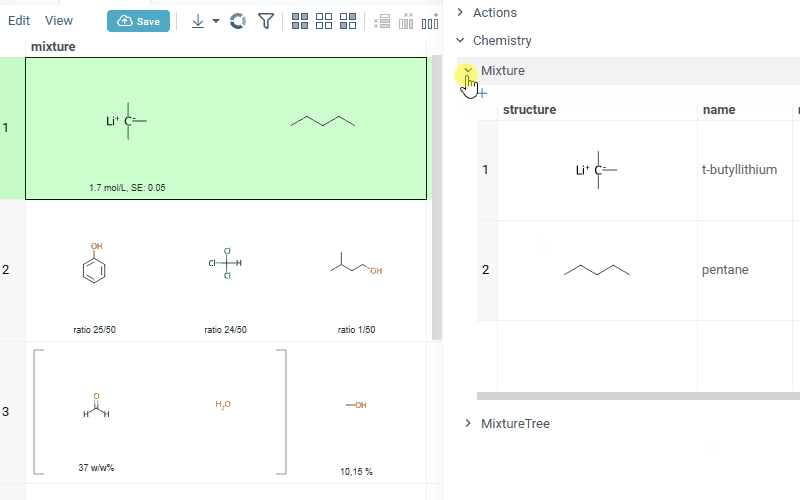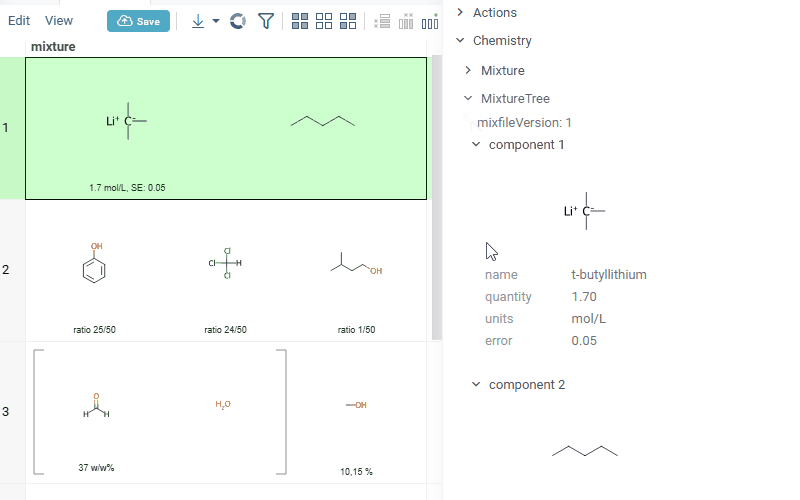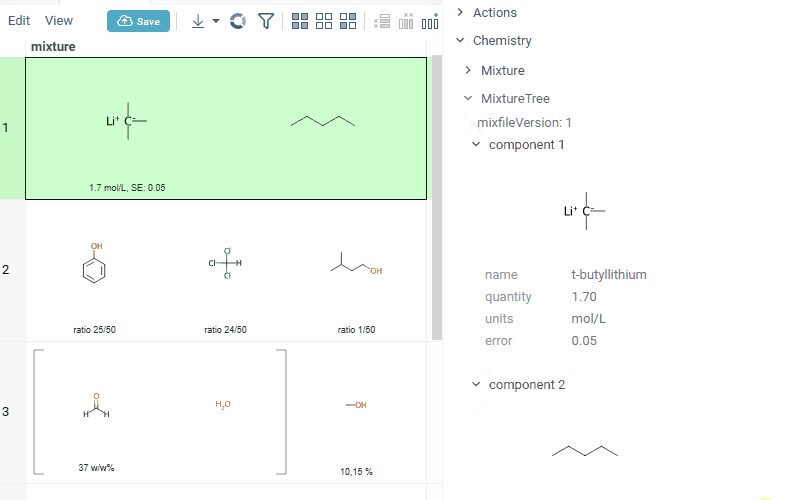
Functionality is available in Chem package version 1.16.0 and upper.
3 Likes
Hierarchical clustering: cluster assignment
You can now generate a cluster column directly from the dendrogram by specifying either a tree height threshold (measured from the root) or the desired number of clusters.
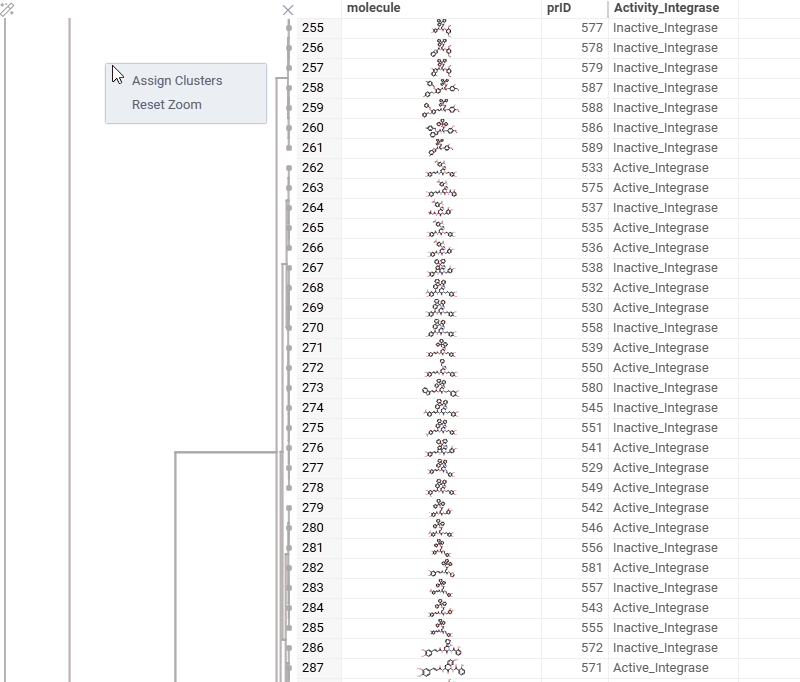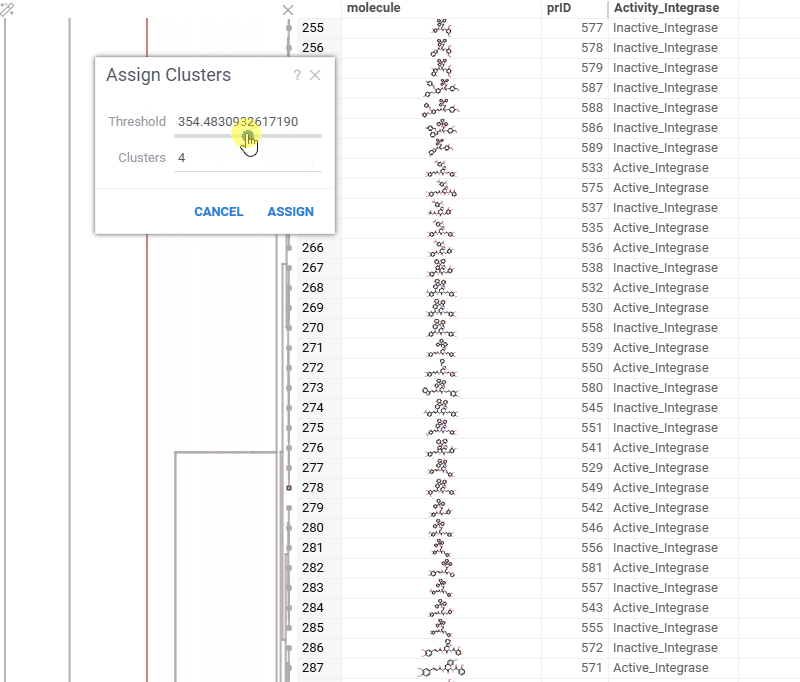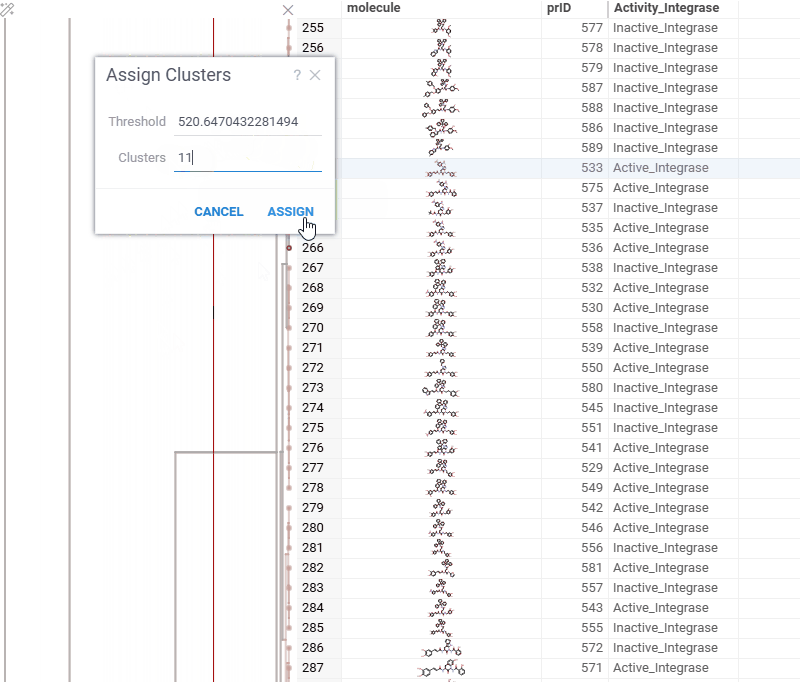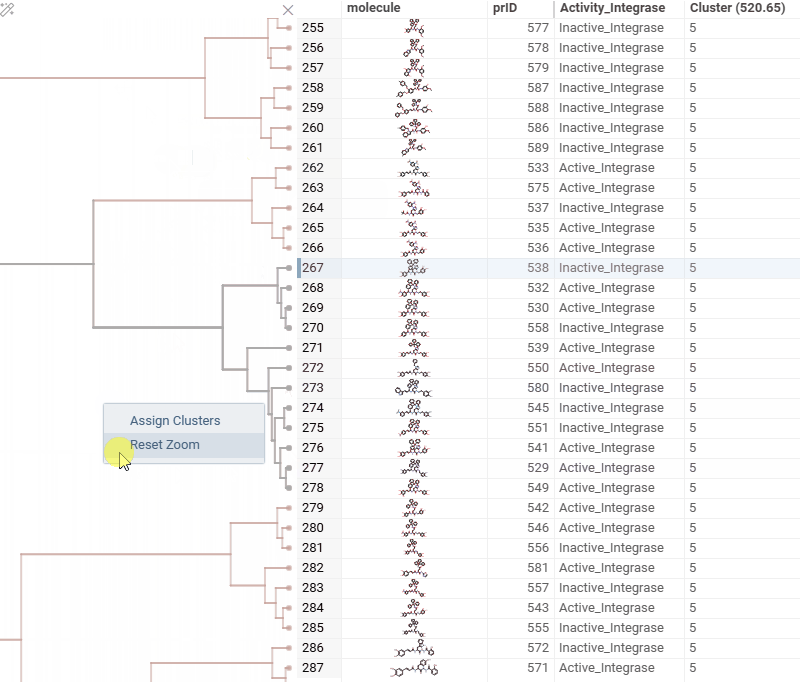
To assign clusters, right-click the dendrogram and select Assign Clusters, or use the Magic Wand icon in the top-left corner of the viewer. In the dialog that opens, specify a threshold or define the number of clusters. The two inputs are interconnected — changing one automatically recalculates the other. For example, if you set the number of clusters to 10, Datagrok will search for the optimal cut position that produces exactly (or as close as possible to) 10 clusters.
The resulting column is named Cluster(thresholdValue), ensuring that the clustering parameters used are preserved and visible in the table.
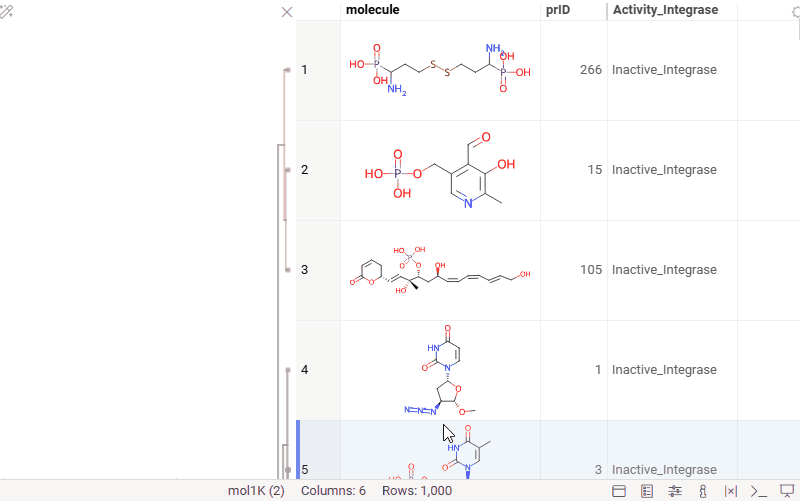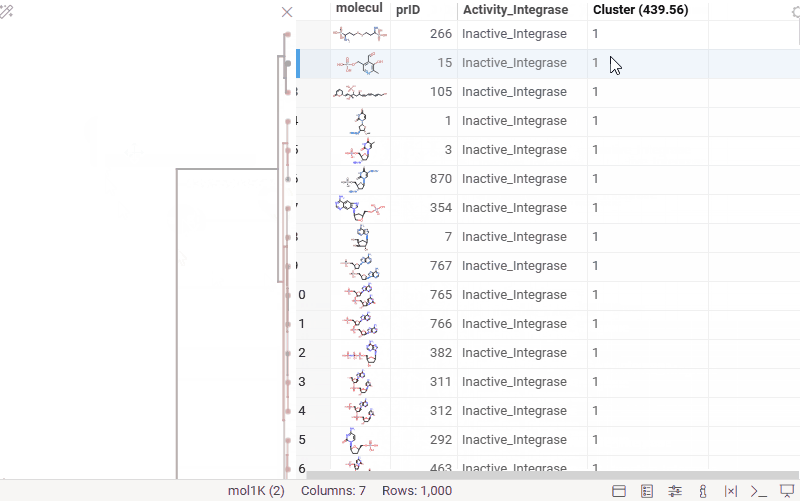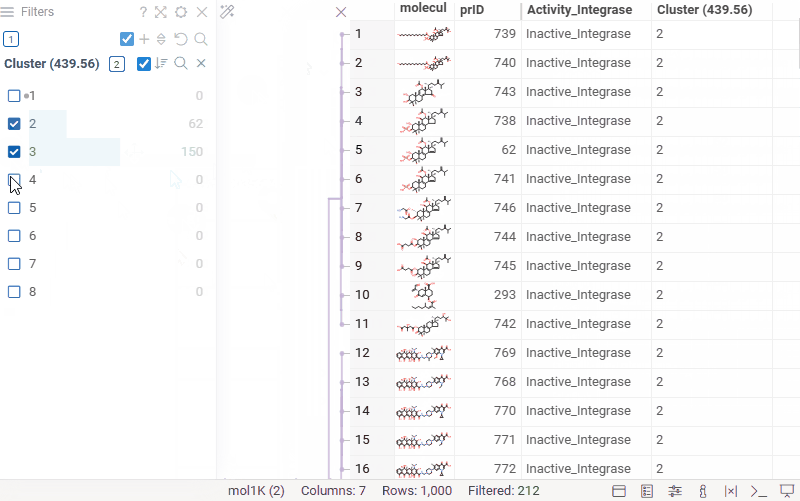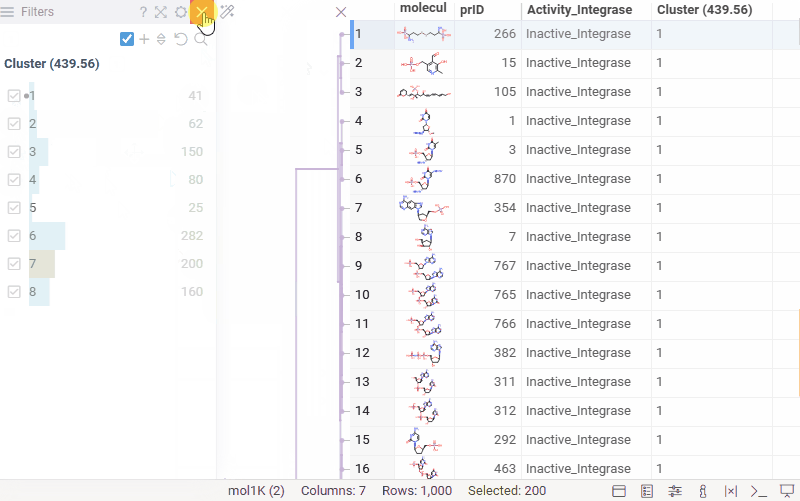
2 Likes
Hierarchical clustering: further improvements
- Dendrogram split visualization: when assigning a threshold or specifying the number of clusters, you can now see exactly where the split occurs on the dendrogram.
- Horizontal zooming: in many cases, clusters are too close together, so now you can zoom in on the X axis using Ctrl + mouse wheel
2 Likes