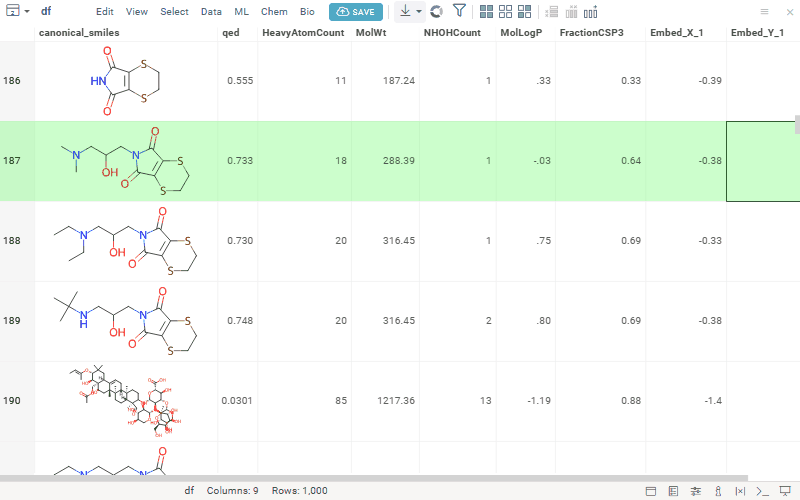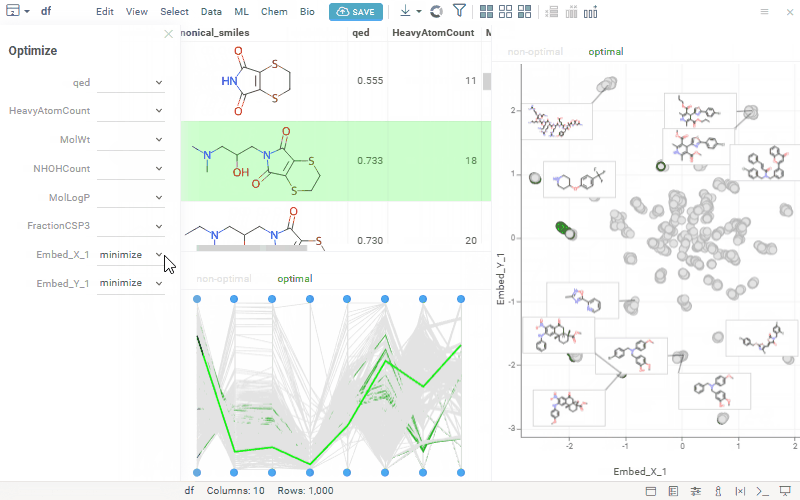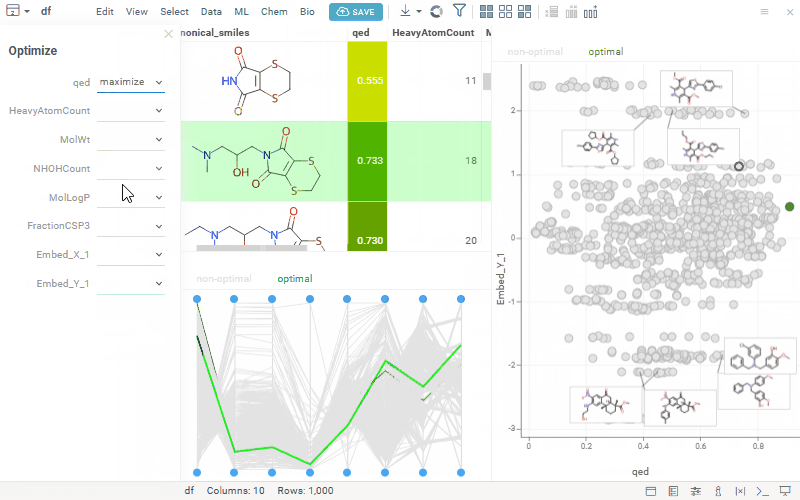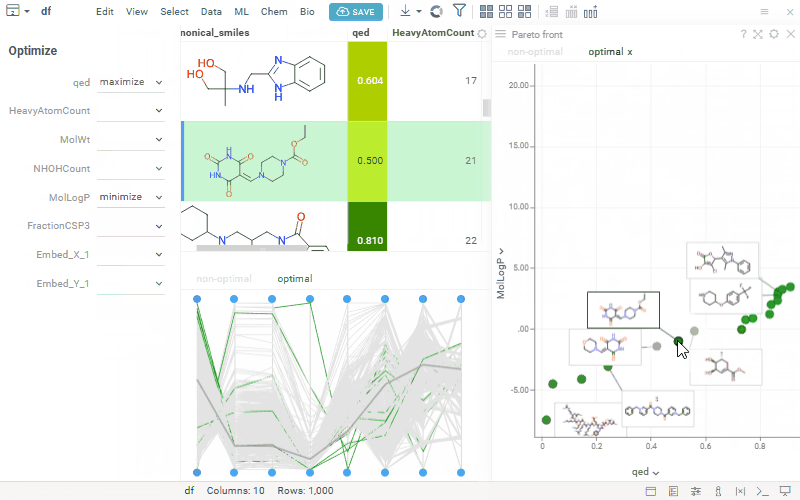
We’ve seen growing interest in our MPO features, so we’re opening a community discussion to learn more about how you’re using them and what you’d like to see next. This is an opportunity to share feedback, align expectations, and collaborate on how MPO can best support your workflows. Here’s a quick reminder of what’s currently available:
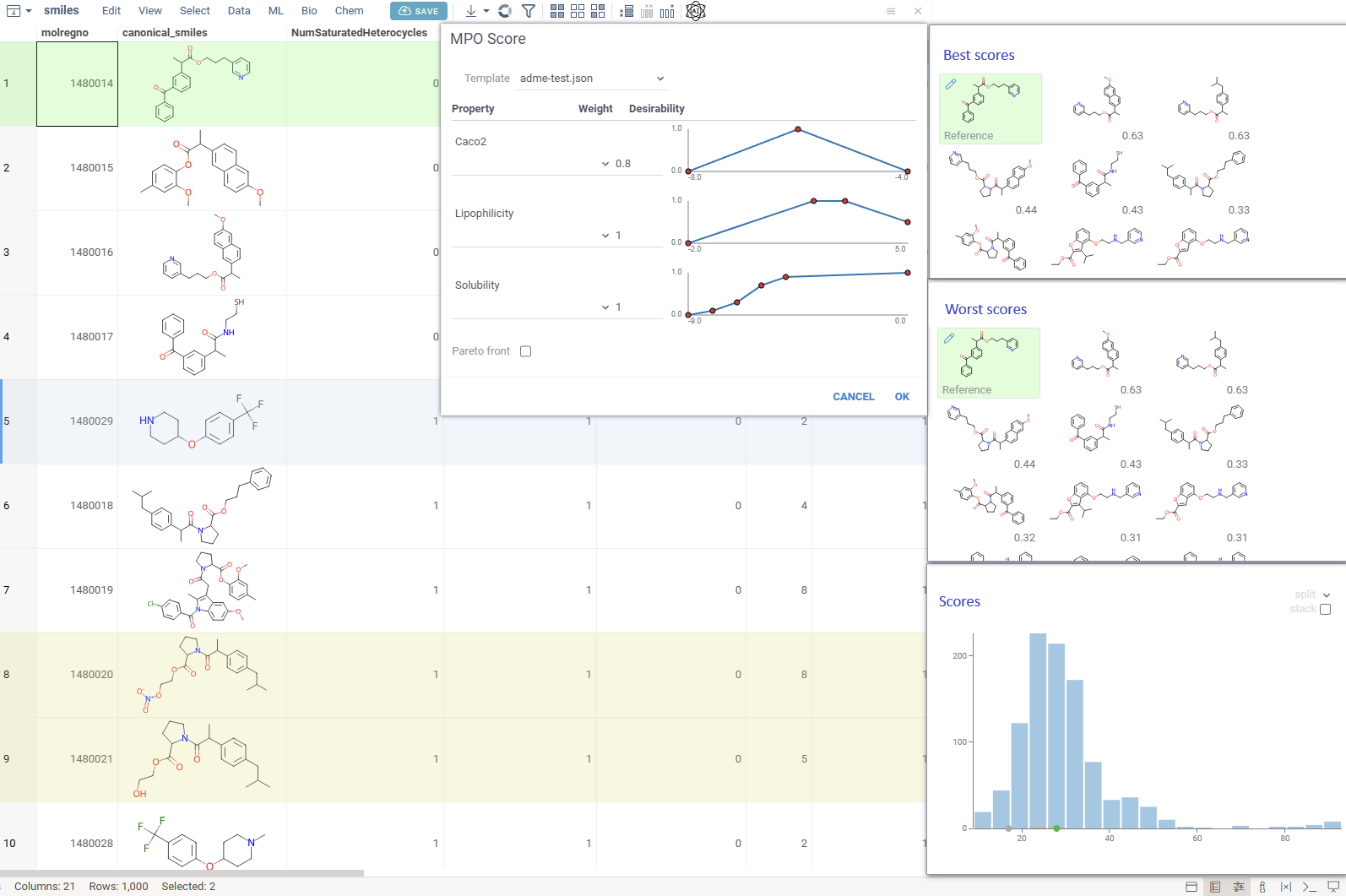
Multiparameter optimization (MPO) helps chemists rank compounds by combining multiple properties — like potency, solubility, permeability, clearance, safety, and selectivity — into a single score, making it easier to prioritize the most promising candidates.
See it in action:
Access MPO via Chem → Calculate → MPO Score… and explore compound profiles interactively.
Main features:
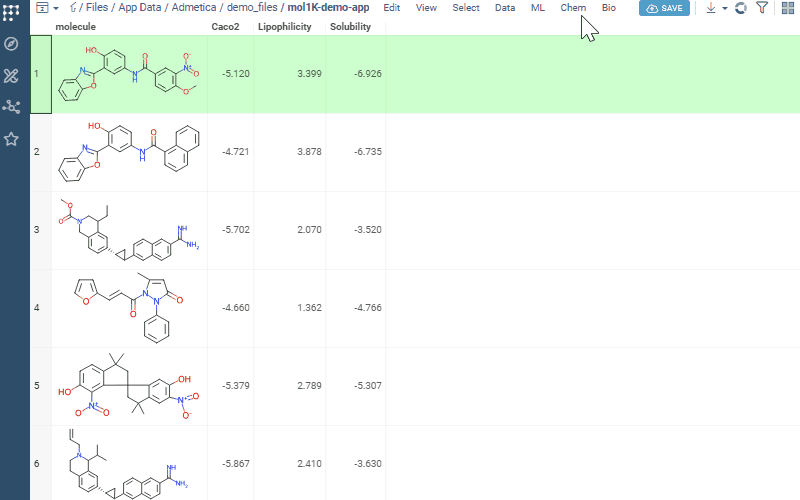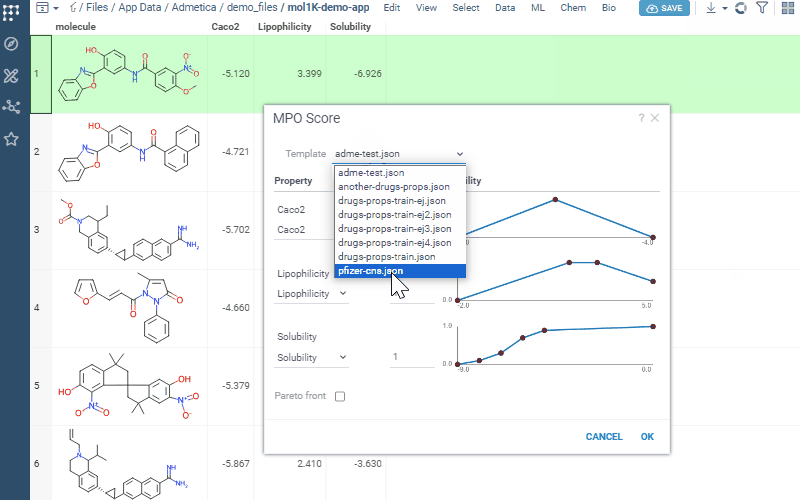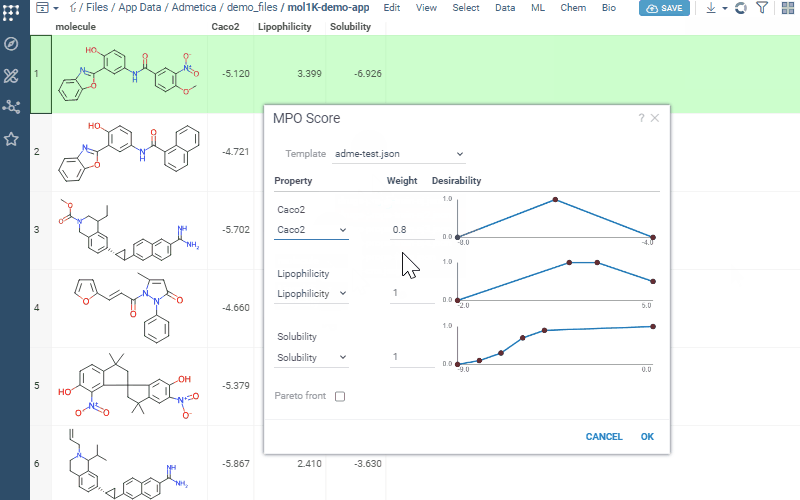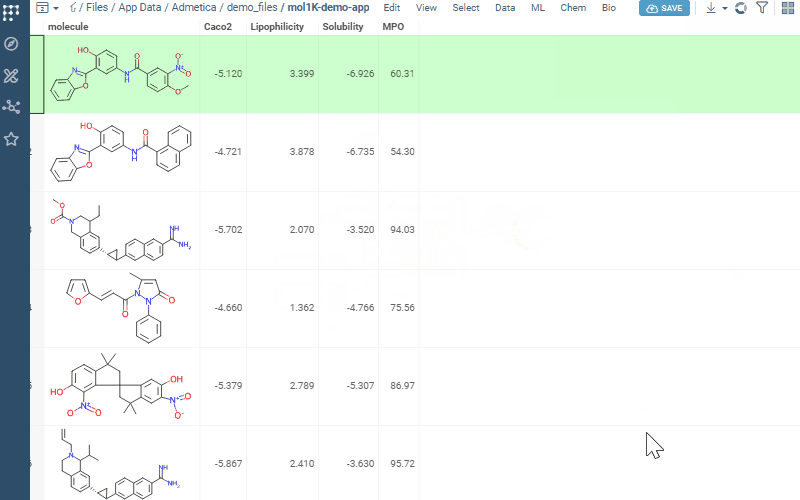
Uses profiles to define included properties, desirability curves, and relative weights
Transforms each property onto a 0–1 desirability scale and aggregates them into a composite score (sum, mean, or weighted)
Includes the built-in Pfizer CNS MPO template, combining six physicochemical properties into a 0–6 score correlated with CNS success
Pareto Front helps chemists identify compounds with the best trade-offs between multiple, often conflicting properties — highlighting non-dominated candidates where improving one objective would worsen another.
See it in action:
Launch via Top Menu → ML → Pareto Front… or add the Pareto Front viewer from the menu ribbon (requires the EDA package).
Main features:
Highlights Pareto-optimal compounds
Supports defining objectives to minimize or maximize selected properties
Allows customization of axes and data point labels
We are interested to hear your ideas on how can Datagrok best support the process of creating the MPO profile. One idea is to provide an interactive feedback on each change. Scores would be recalculated whenever any change is made to the profile (added or removed properties or changed the desirability function), but in addition to that we are thinking of providing this information in the context panel:
Compounds with the best MPO scores
Compounds with the worst MPO scores
Distribution of the MPO scores
Would that be useful? What else can we do to better guide the MPO profile design?
Thanks for these comments. We fully agree that editing existing profiles is the top priority. Coverage for categorical properties and default scores definitely on the short term roadmap. Your cascade idea is interesting. How do you ensure that fall-back properties are in the same scale as the primary properties? How frequently is cascading occurring in practice?
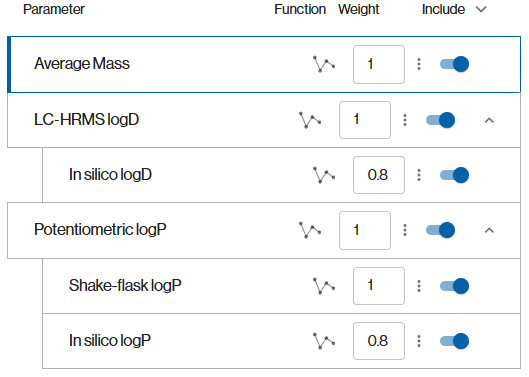
@ejaeger1 Better said “fallback desirability function” - each property has its own desirability function, but some property get considered only if a “parent property” is missing. For instance in this screenshot, if there is no experimental value for Potentiometric logP, the value from the shake-flask experiment will be used. If this one is not available it will use the desirability function of the in silico logP with a lower weight.
Ok. So the story here is the creation of a template that can be used in multiple datasets. You, as the MPO score designer, want to be prepared that different datasets will have alternative properties/measures for the logP parameter and you have a preference as to which should be invoked. So this is a predefined preferential mapping of potential columns and their desirability functions to the parameter of interest in the score.
The bell shape (gaussian) distribution is part of the immediate plan along with a sigmoid distribution augmenting the existing freeform distribution definition.
Log scale transformation is interesting. Not wanting to open pandora’s box, but what other dynamic transformation would be likely. I can see “p” (-log) as relevant.
We would also need the ability to specify arbitrary functions as “parameters” - so that you can run MPO just on the molecules alone, and the necessary properties are calculated automatically if not present in the dataset.
Hi everyone! Thank you for all the feedback you shared during the MPO development — it really helped us shape the tool into what it is today. Here’s what we’ve built:
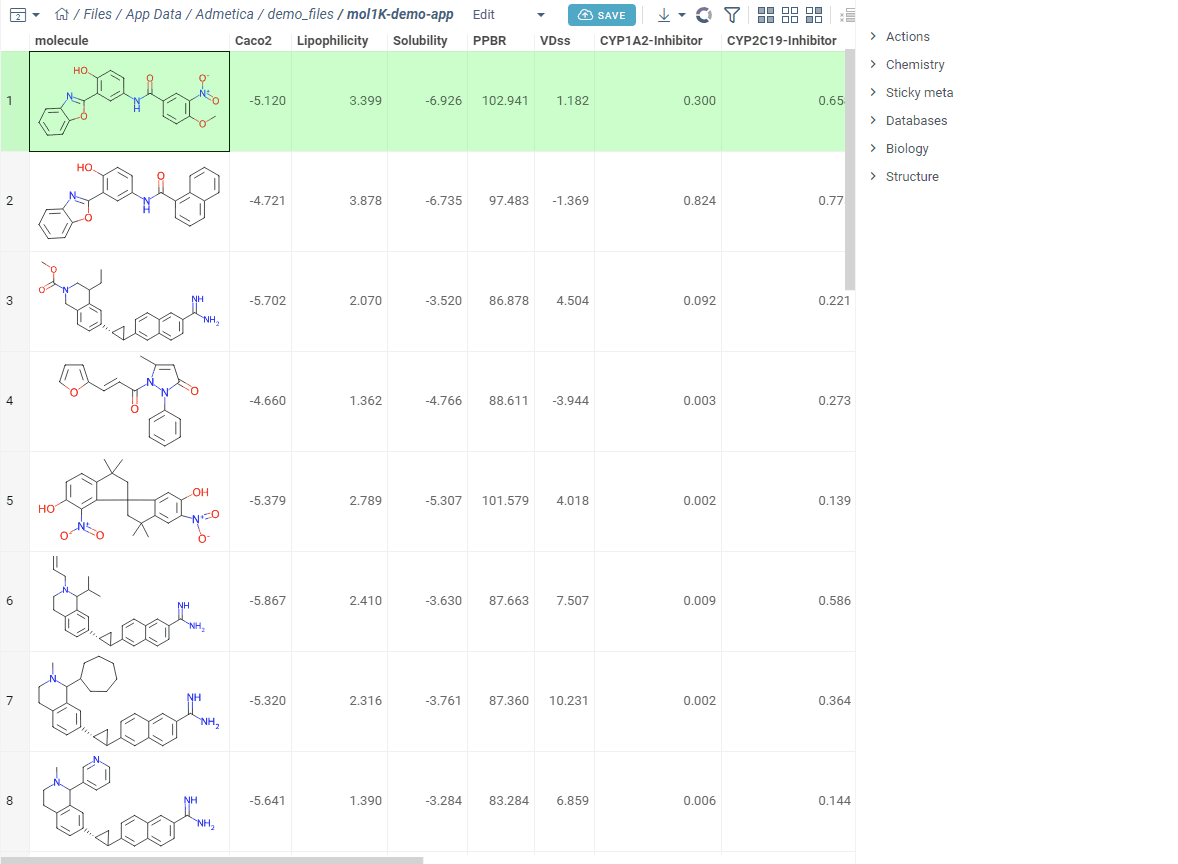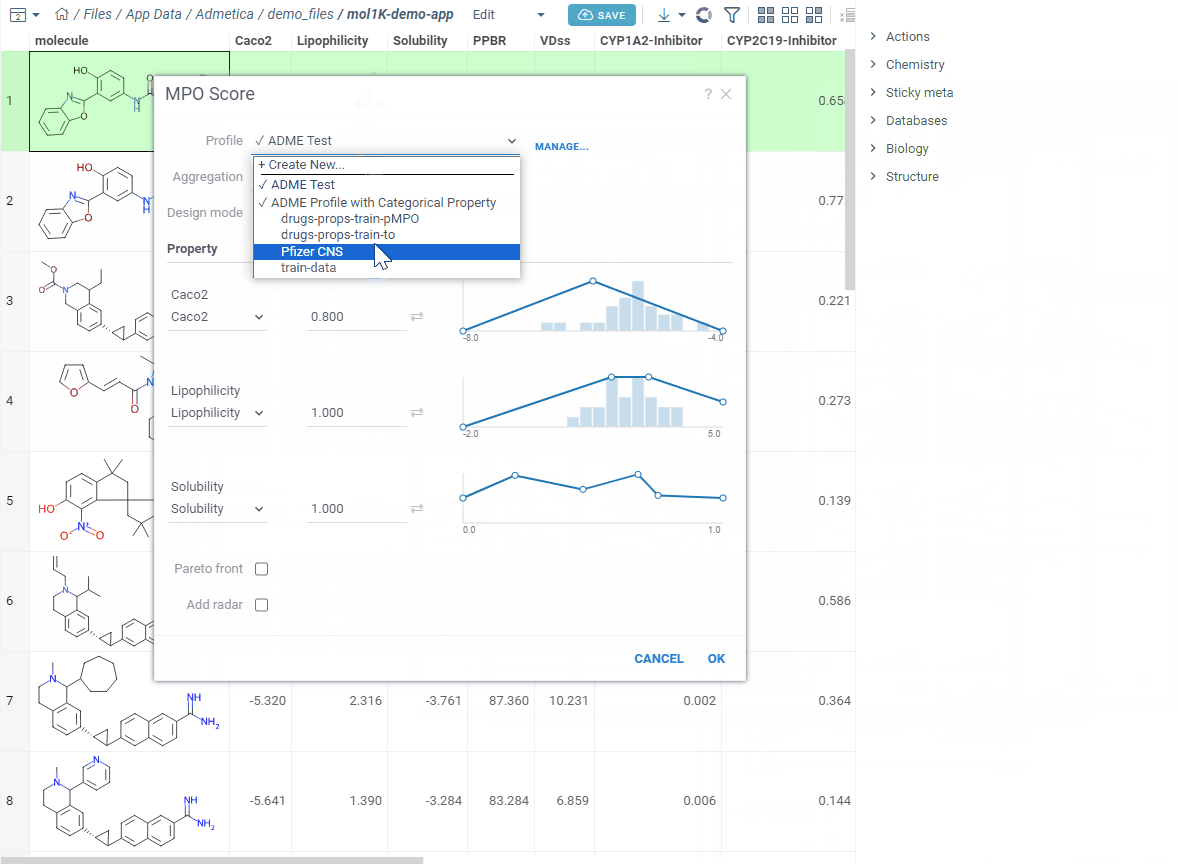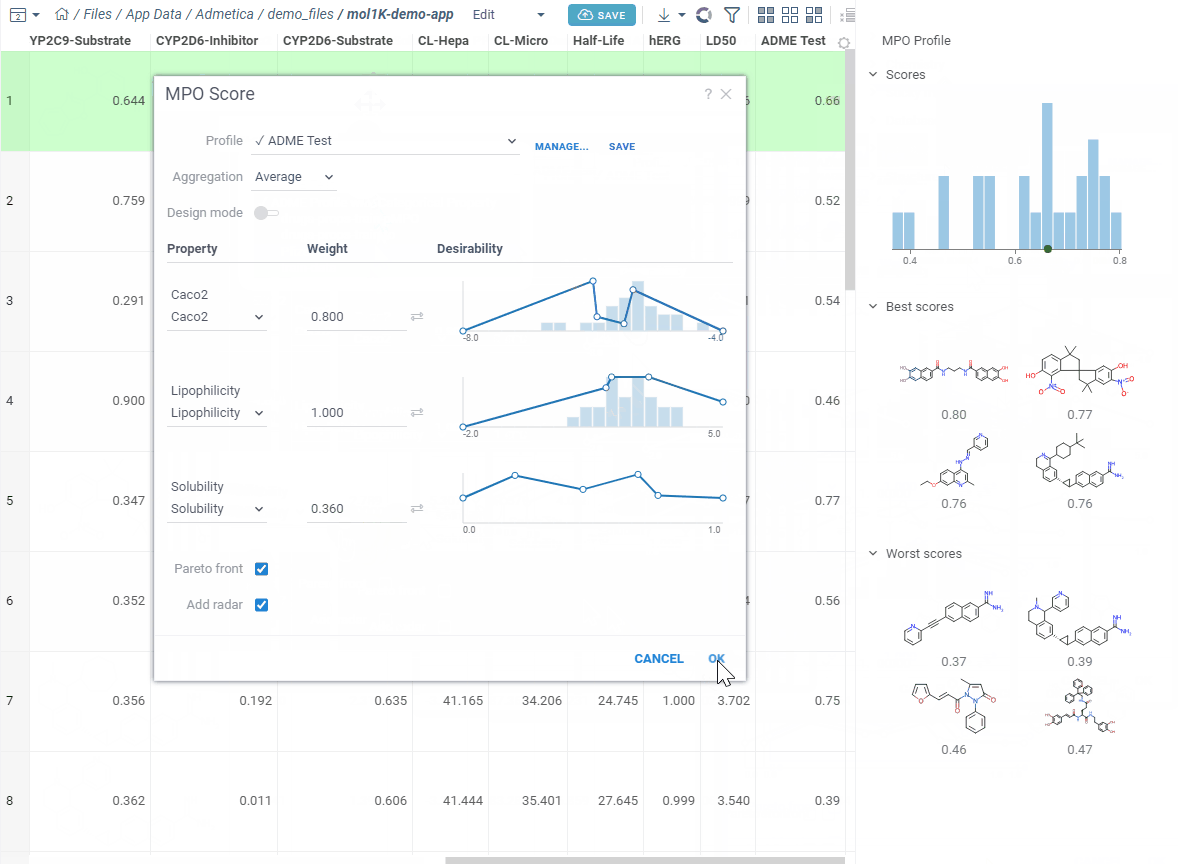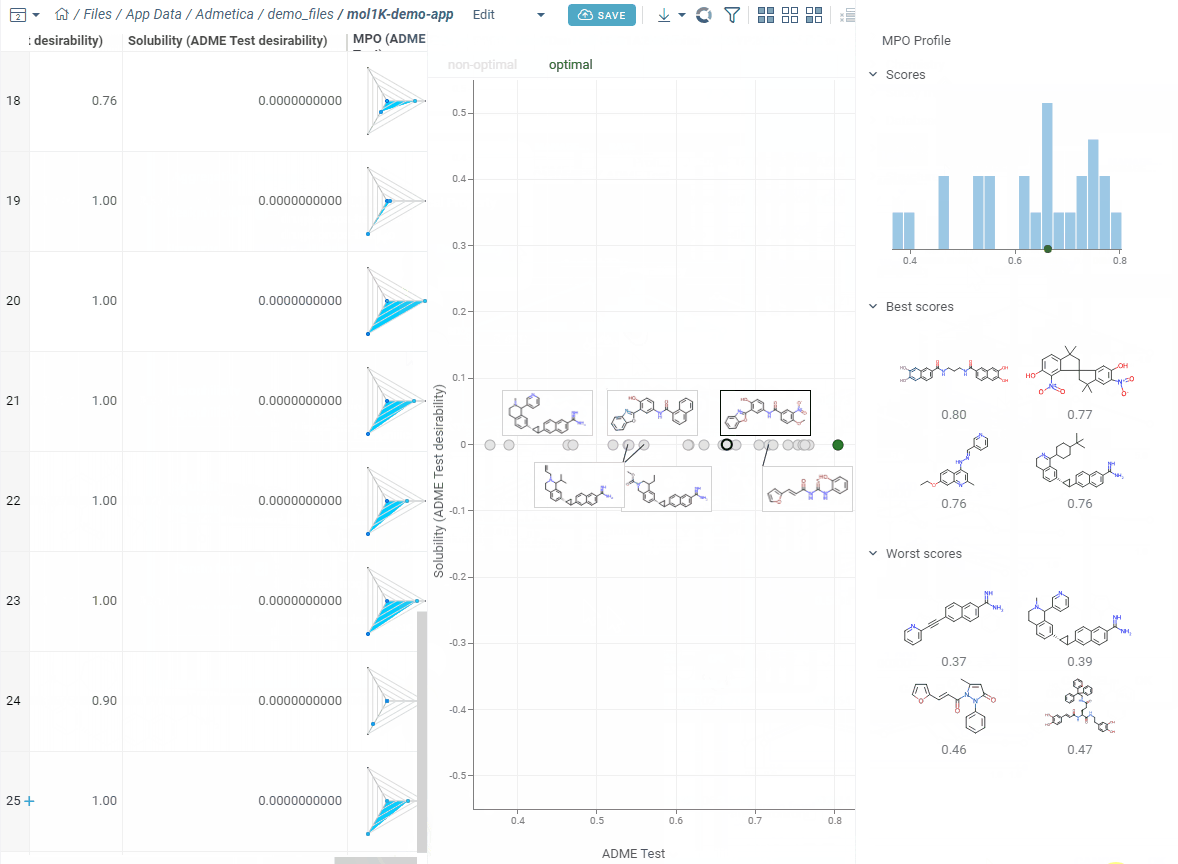
Your starting point is Apps→ Chem → MPO Profiles. Here you see all your saved profiles. You can edit, clone, delete, or download any of them as a JSON file. Someone sent you theirs? Just upload it.
Let’s create a new one — click Create Profile. The editor opens in a split view — curves on the left, your data on the right. Add the properties you care about and start shaping your desirability curves.
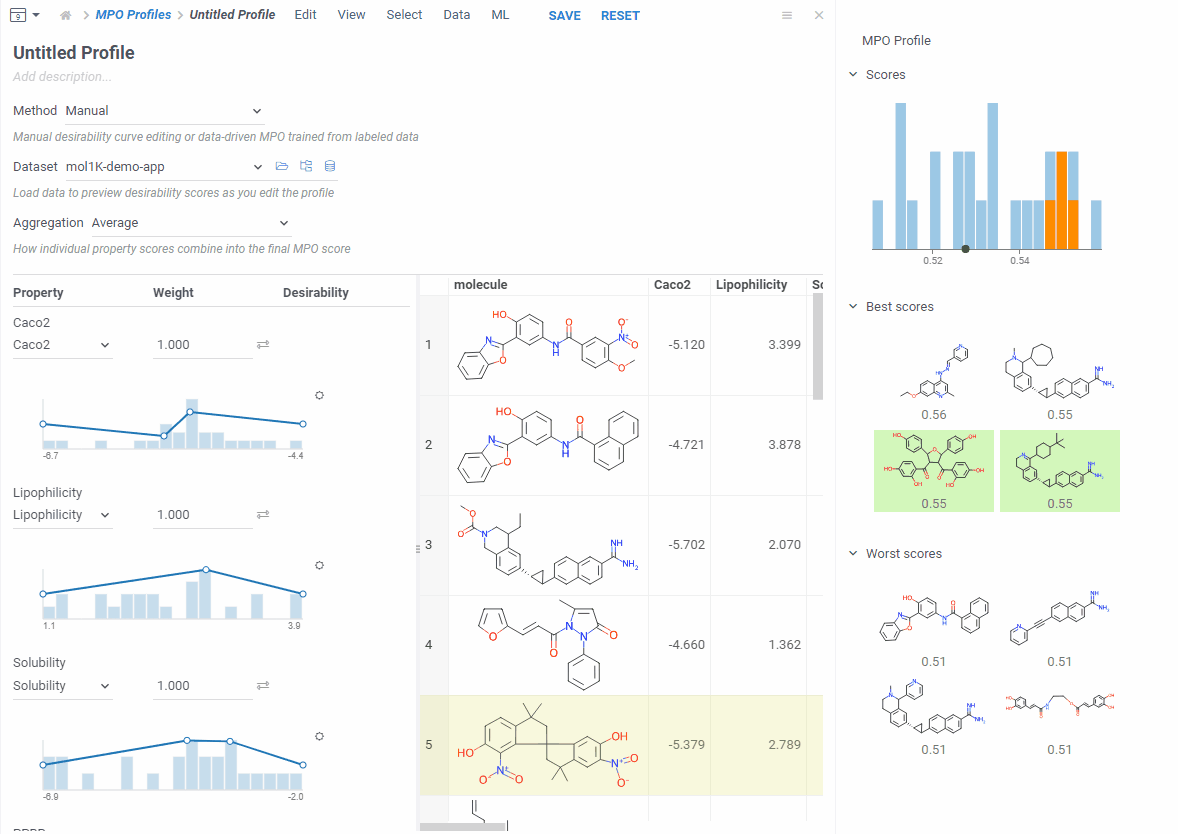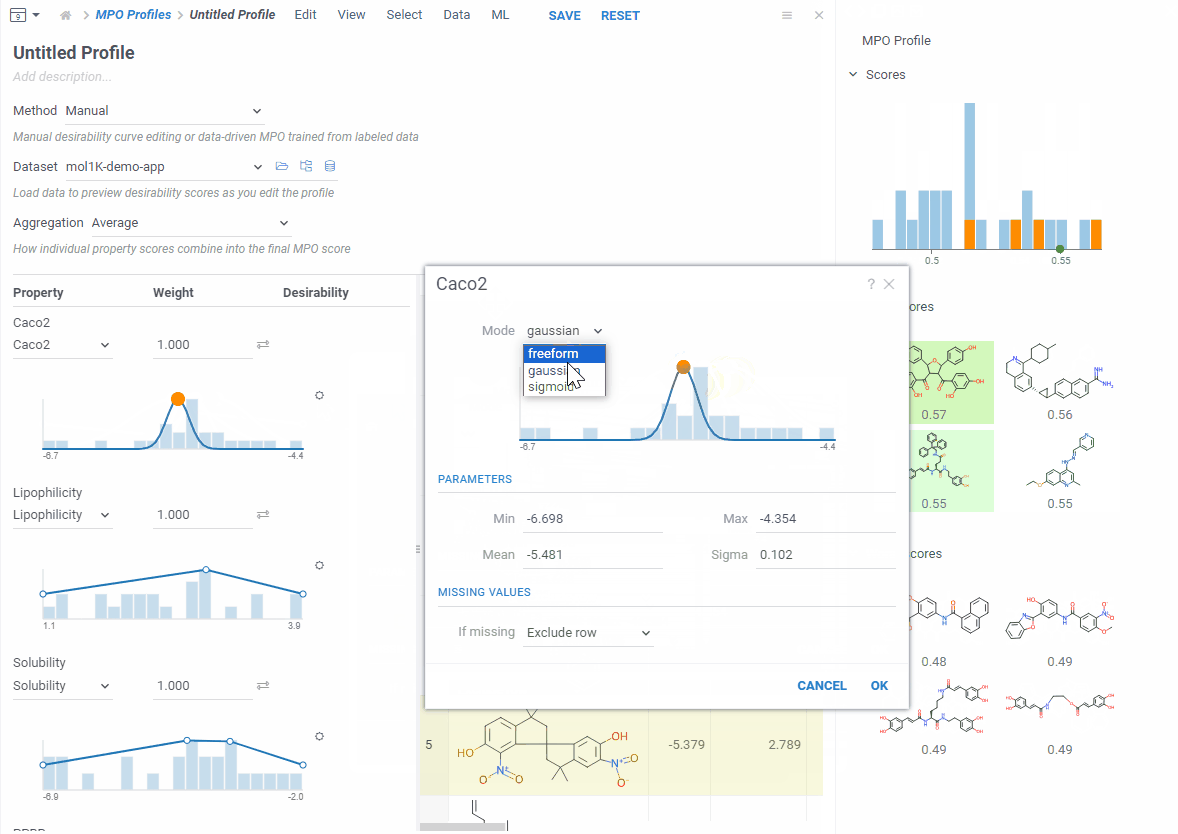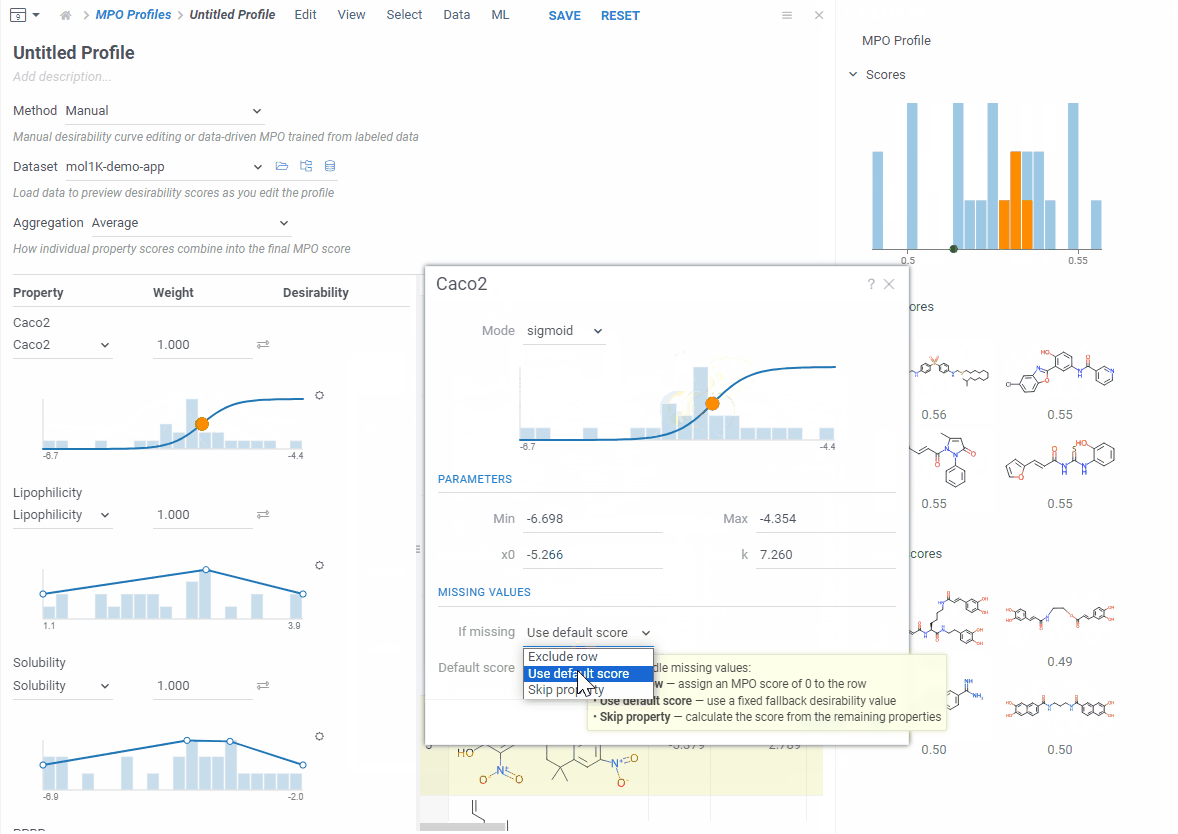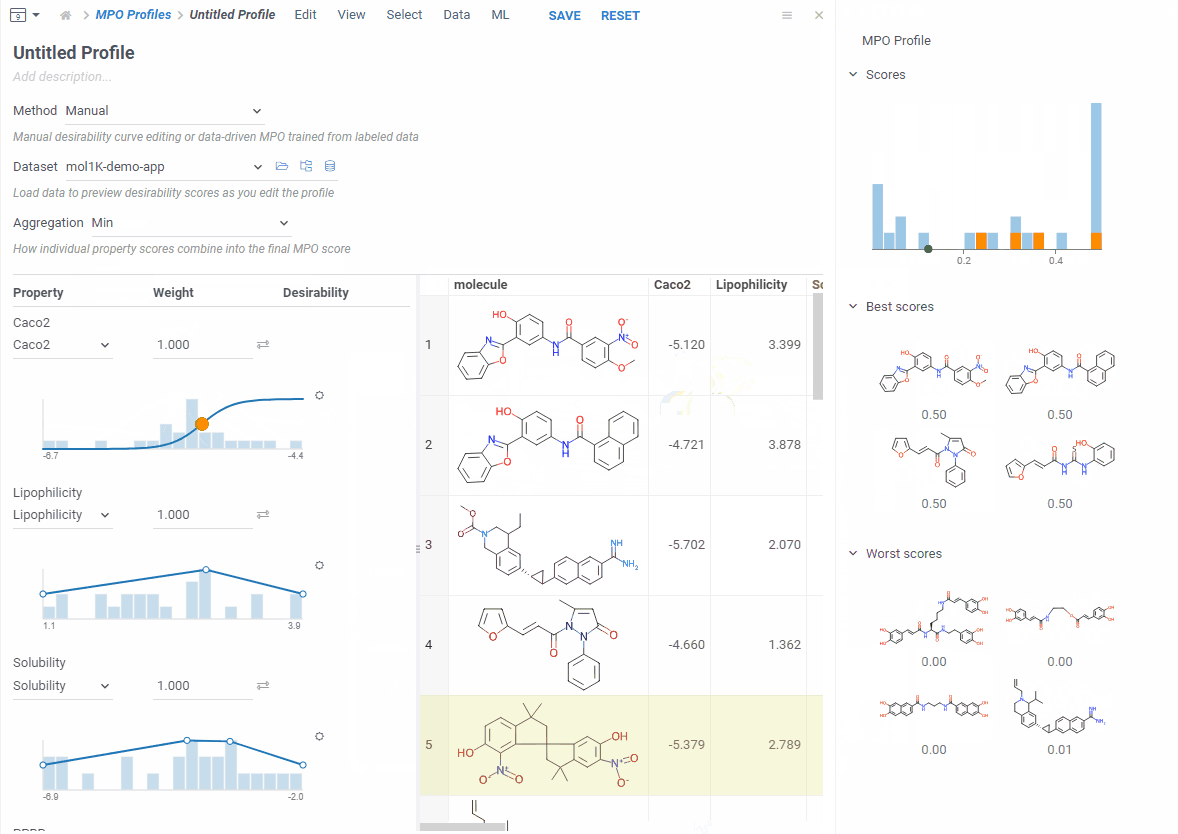
As you edit, the right side updates right away: you see a score histogram and the best and worst scoring molecules. Click any molecule to find it in the grid. This makes it easy to experiment — change a desirability curve, see the effect instantly.
If a property isn’t in your dataset, you can add it as a computed function and MPO will calculate it for you.
Each desirability curve can be one of three types: freeform (draw it yourself), gaussian (bell curve), or sigmoid (S-curve). Open the settings to switch and adjust.
Not every property is a number — for categorical ones, just assign a score to each category. If some values are missing, you choose what to do per property: skip the row, use a fallback score, or ignore that property for that row.
Pick how to combine everything — average, sum, product, geometric mean, min, or max — adjust the weights, and save.
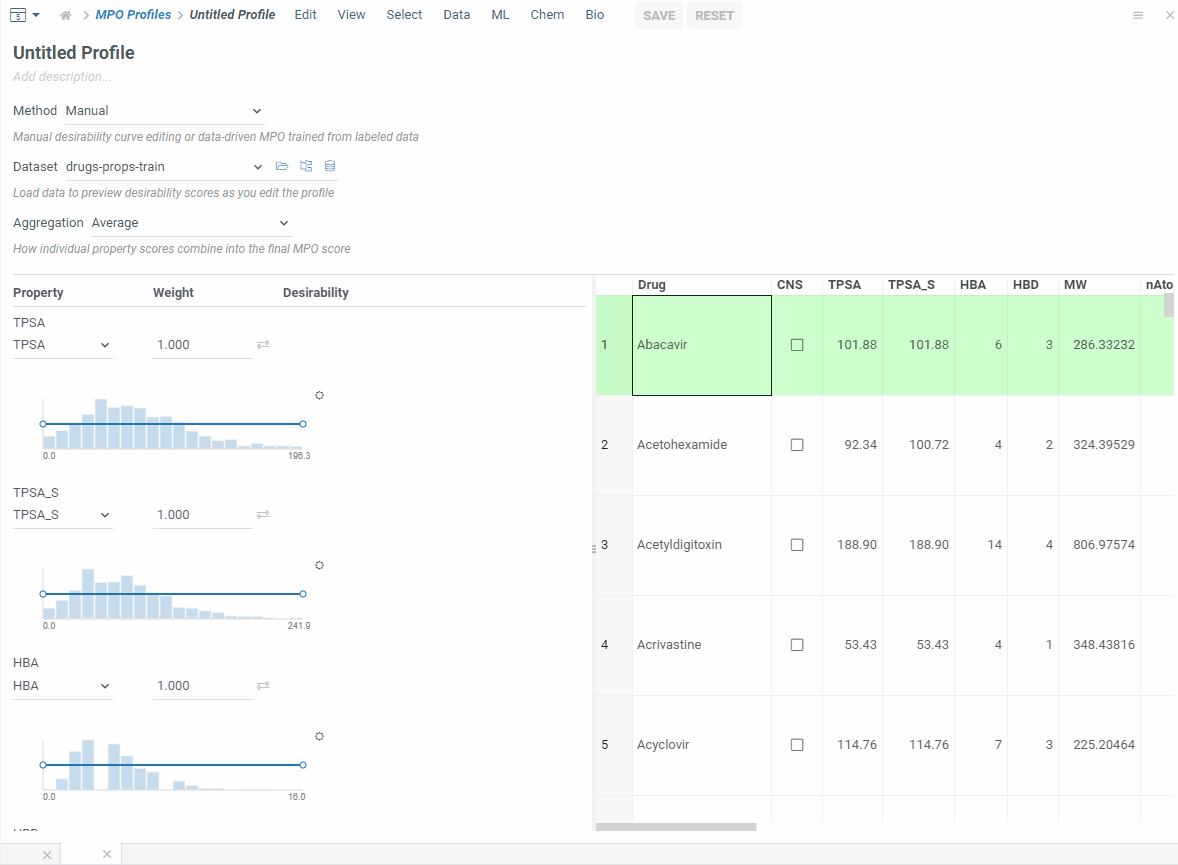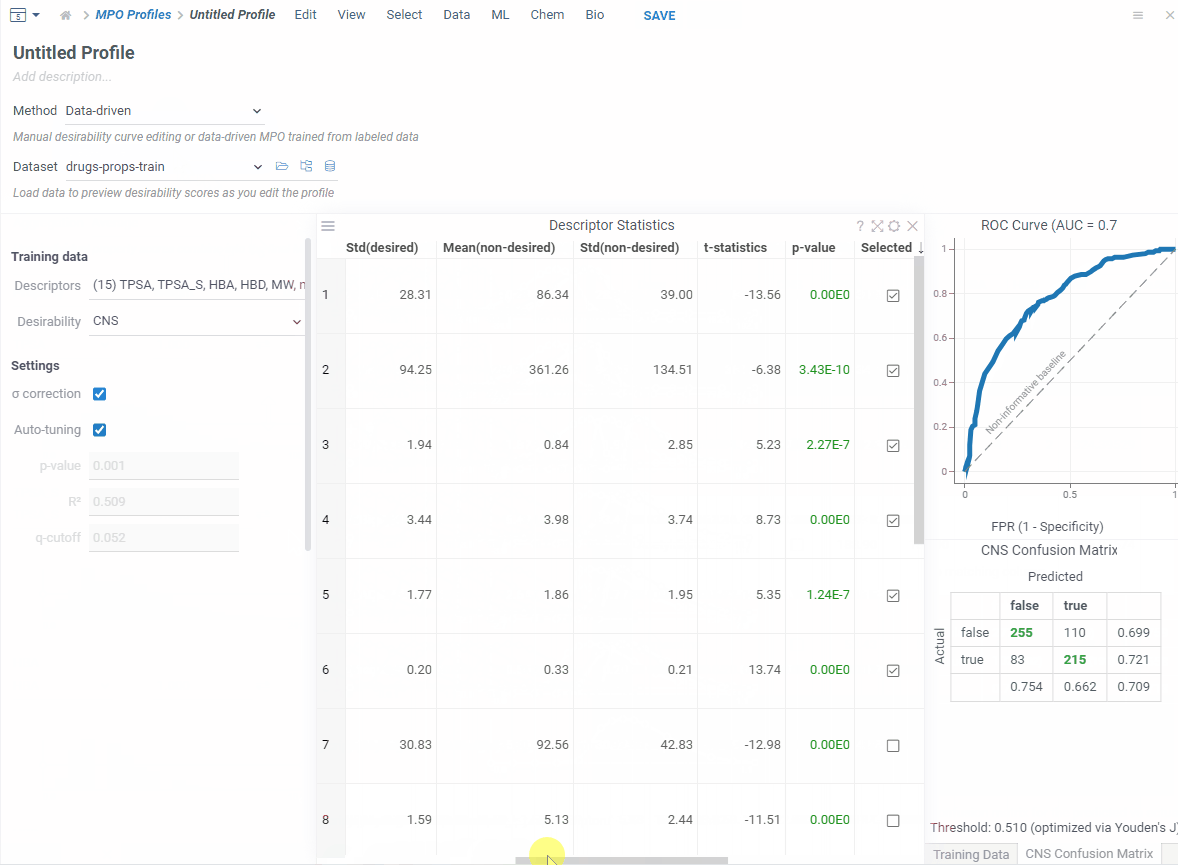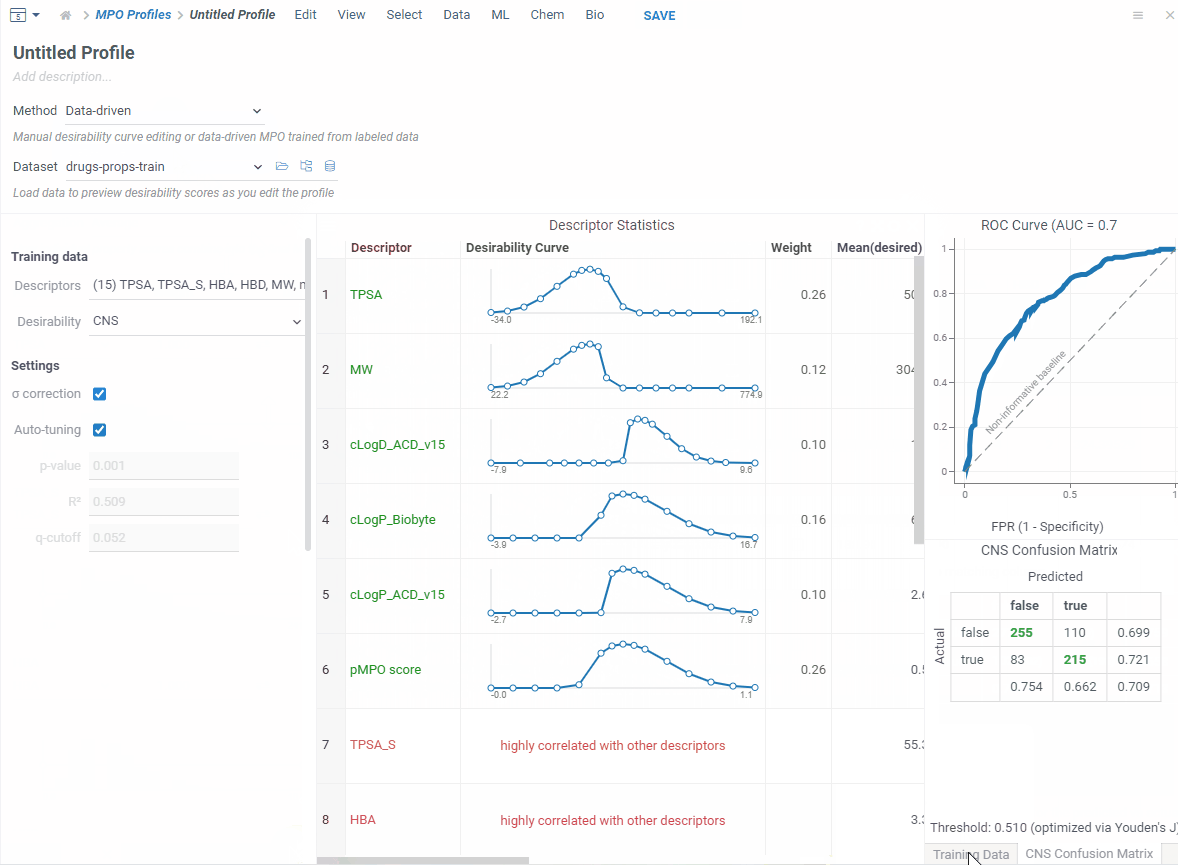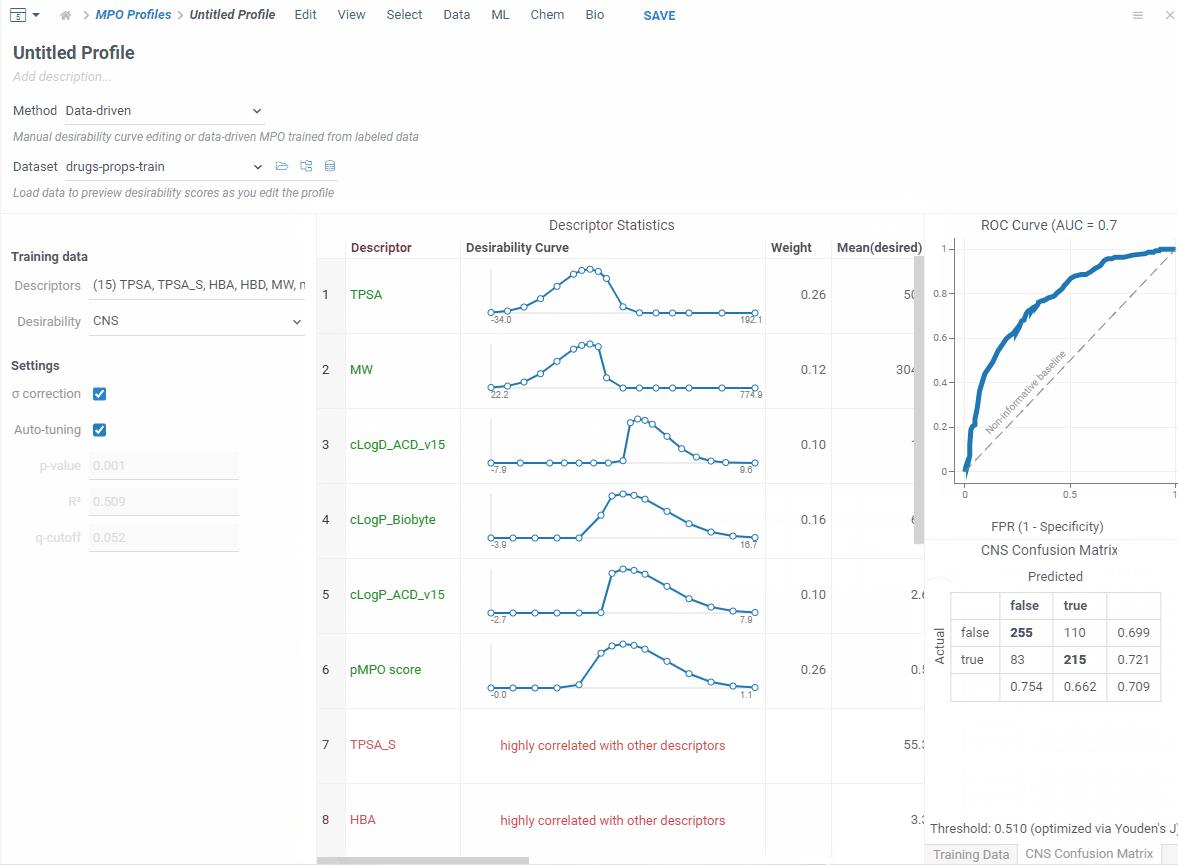
If you have a labeled dataset you trust, switch to data-driven mode. Your data already captures what makes a good profile — so MPO can build one directly from it.
Pick a column that defines what’s desirable — boolean, numeric or categorical — and MPO will shape everything automatically!
In addition, you get a ROC curve and confusion matrix to check how well it performs before saving.
Now let’s score. Open your dataset and go to Chem → Calculate → MPO Score. Profiles that match your columns show a ✓ — pick one.
Turn on Pareto Front to spot molecules with the best trade-offs between competing properties. Add Radar Chart to see each molecule’s score breakdown right in the grid. Sort by MPO score and you’re looking at your top candidates.